O PÁGINA UM analisou, com detalhe, e com os dados possíveis, a evolução da agressividade da covid-19 em Portugal desde o início da pandemia. E apurou que as taxas de internamento e de letalidade global agora com a variante Ómicron a dominar são já inferiores às que se registam em surtos gripais em países com estimativas para aquela doença, como os Estados Unidos. Só o risco global de morte para o pequeno grupo dos que são internados por covid-19 ainda continua a superar o da gripe, mas tal dever-se-á aos grupos vulneráveis. Apesar de haver muitos que insistem numa alegada 6ª vaga para vender antivirais experimentados com variantes mais agressivas, a pergunta coloca-se: vale a pena tal esforço financeiro quando o SARS-CoV-2 se mostra agora muito mais “sereno”? E mais outra: não há mais prioridades em Saúde Pública?
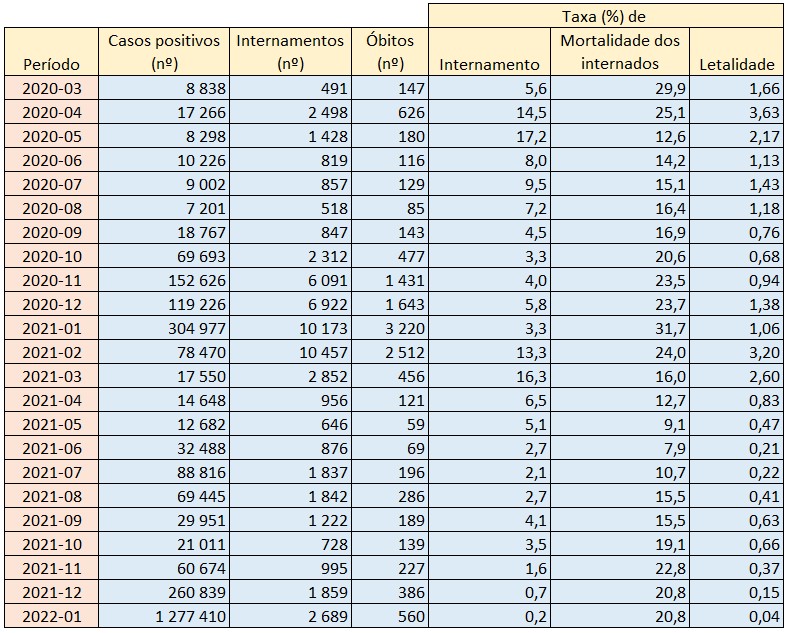
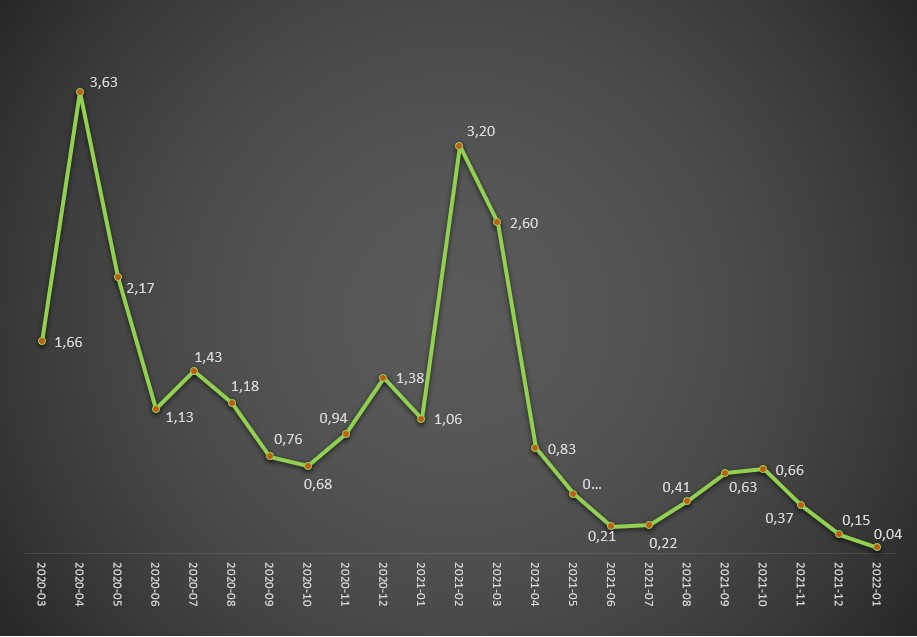
Em Janeiro deste ano, a taxa de internamento de infectados com o SARS-CoV-2 foi de apenas 0,2%, e a taxa de letalidade da covid-19 situou-se somente em 0,04%, os valores mais baixos desde o início da pandemia. Ou seja, em cada 1.000 casos positivos detectados no primeiro mês de 2022 somente duas pessoas acabaram internadas.
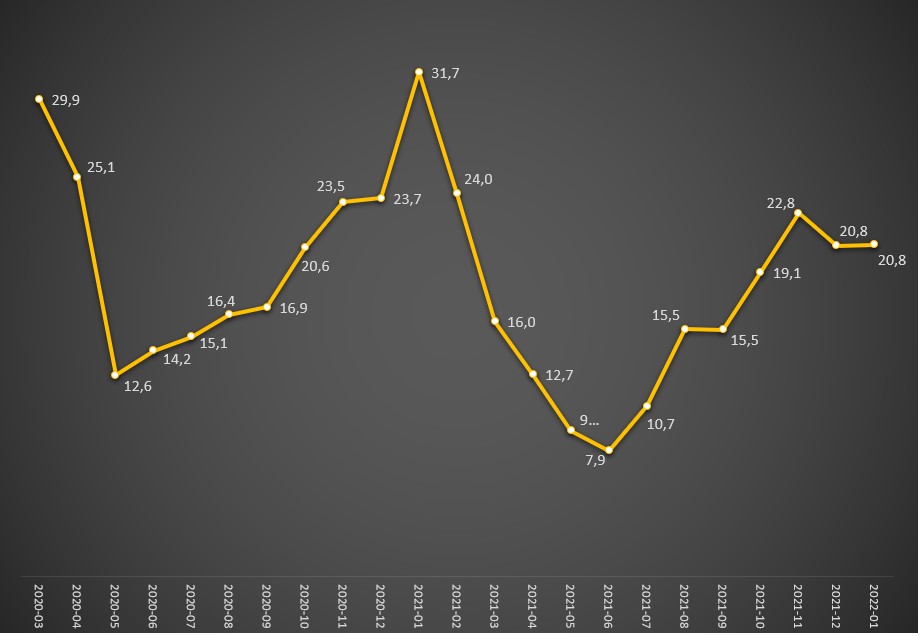
Como o risco de morte dos internados rondava então os 21%, significa que no primeiro mês deste ano, que correspondeu até a uma elevada incidência, morreu uma pessoa por cada 2.500 casos positivos. No período de maior agressividade da pandemia, a covid-19 chegou a apresentar uma taxa de letalidade global de 3,2% (Fevereiro de 2022), considerando os óbitos registados nos hospitais, ou seja, 16 vezes superior. Portanto, naquele mês, para cada 2.500 casos positivos houve 16 óbitos.
Estas são as principais conclusões de uma análise exclusiva do PÁGINA UM, através do cruzamento dos casos positivos por mês, divulgados pela Direcção-Geral da Saúde (DGS), com a base de dados da morbilidade e mortalidade hospitalar do Portal da Transparência do Serviço Nacional de Saúde (SNS).
Saliente-se que, no caso dos óbitos, estão apenas incluídos os óbitos por covid-19 com registo em unidades do SNS. O Ministério da Saúde nunca esclareceu a razão pela qual cerca de um terço das vítimas do SARS-CoV-2 – que acabaram por morrer com graves insuficiências respiratórias – terem falecido sem tratamento hospitalar.
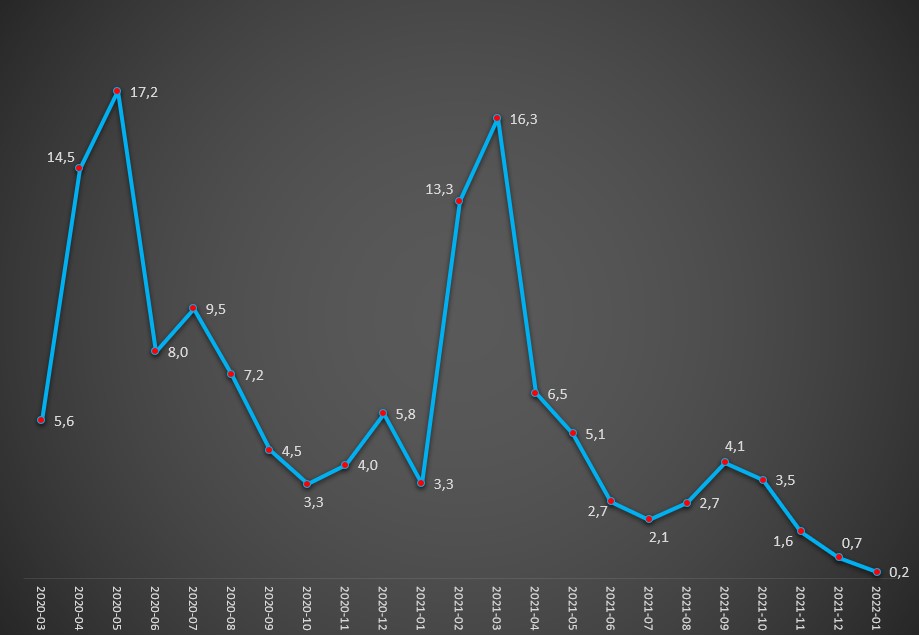
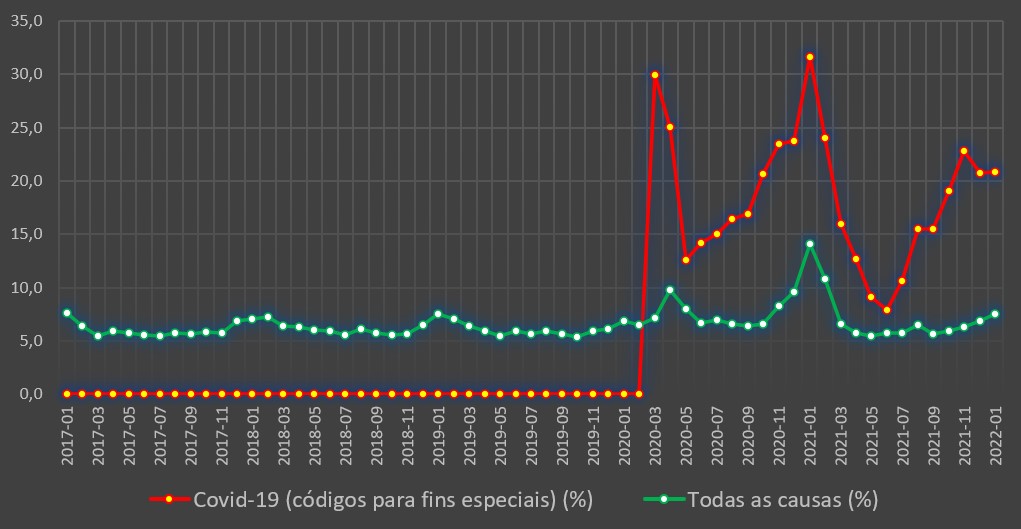
Mostra-se, em todo o caso, evidente que, apesar do surgimento da variante Omicron ter provocado uma subida abrupta de casos positivos, a agressividade do covid-19 decaiu significativamente. Nas fases de dominância das variantes Alfa (Primavera de 2020) e Delta (primeiros meses de 2021), as taxas de hospitalizações chegaram a rondar, ou estar mesmo acima, dos 15%. Ou seja, por cada 1.000 casos positivos, 150 acabavam por ser hospitalizados.
Número de casos positivos, internamentos, óbitos atribuídos à covid-19 e respectivas taxas (%) de internamento, mortalidade dos internados e letalidade global em Portugal por mês. Fonte: DGS / Worldometers e SNS. Cálculos e análise: PÁGINA UM.
Em Janeiro do ano passado – o mês com maior número de mortes atribuídas à covid-19 –, a taxa nem esteve exageradamente alta (3,3%), mas devido ao colapso do SNS e à vaga de frio a taxa de mortalidade hospitalar por esta doença atingiu um pico de quase 32%, como revelou o PÁGINA UM na semana passada.
Contudo, desde o surgimento e dominância da variante Omicron, no final do ano passado, a taxa de hospitalizações por covid-19 começou a cair abruptamente. Em Novembro de 2021 foi de 1,6% (16 internamentos em cada 1.000 casos positivos), o que já era o valor mais baixo de sempre. Em Dezembro desceu para 0,7% (7 internamentos em cada 1.000 casos positivos) e em Janeiro passado – últimos dados disponíveis – já somente atingiu os 0,2%.
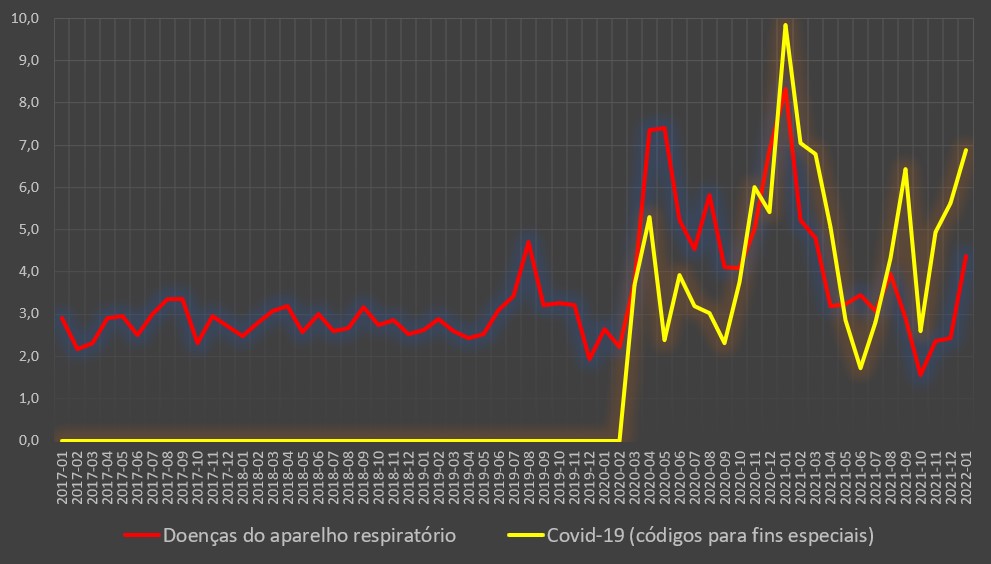
Evolução da taxa (%) de internamento atribuída à covid-19 (internados por casos positivos) entre Março de 2020 e Janeiro de 2022. Fonte: DGS / Worldometers e SNS. Cálculos e análise: PÁGINA UM.
Apenas uma análise mais fina, estratificada por grupos etários, permitiria apurar se esta diminuição abrupta foi homogénea para toda a população ou se se verificam diferenças distintas em função da idade.
Porém, apesar desses elementos serem recolhidos e tratados pelo Sistema Nacional de Vigilância Epidemiológica (SINAVE), a DGS tem manifestado uma sistemática atitude obscurantista, razão pela qual o PÁGINA UM intentou na semana passada um processo de intimação junto do Tribunal Administrativo de Lisboa contra o Ministério da Saúde. Uma das bases de dados que o PÁGINA UM pretende aceder é, exactamente, o SINAVE.
Evolução da taxa (%) de mortalidade hospitalar dos internados com covid-19 (óbitos por internados) entre Março de 2020 e Janeiro de 2022. Fonte: DGS / Worldometers e SNS. Cálculos e análise: PÁGINA UM.
Em todo o caso, mesmo com base nos dados globais, do ponto de vista epidemiológico os indicadores da covid-19 começam, cada vez mais, a assemelhar-se a um surto gripal. Com efeito, embora em Portugal não existam sequer estimativas razoáveis sobre a incidência da gripe, a taxa de hospitalização e mortalidade associada ao vírus influenza (também como “porta de entrada” das subsequentes pneumonias), indicadores dos Estados Unidos permitem uma comparação razoável.
Evolução da taxa (%) de letalidade atribuída à covid-19 (mortes nos hospitais por casos positivos) entre Março de 2020 e Janeiro de 2022. Fonte: DGS / Worldometers e SNS. Cálculos e análise: PÁGINA UM.
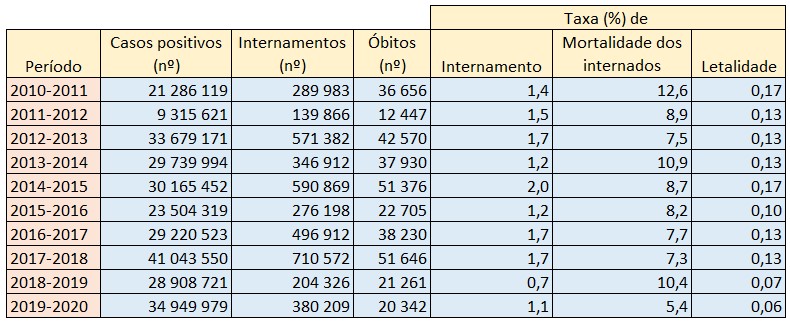
Com efeito, de acordo com as estimativas anuais do Centers for Disease Control and Prevention (CDC), nas épocas de 2010-2011 a 2019-2020, a taxa de internamento associado à gripe situou-se entre os 0,7% (2018-2019) e os 2,0% (2014-2015), enquanto a taxa de letalidade esteve compreendida entre os 0,06% (2019-2020) e os 0,17% (2014-2015).
Porém, a taxa de mortalidade hospitalar no caso das gripes mostra-se, em comparação com a situação dos internados-covid em Portugal (que ronda os 20%), substancialmente menor, situando-se entre os 5,4% (2019-2020) e os 12,6% (2010-2011).
Número de casos positivos, internamentos, óbitos atribuídos à covid e respectivas taxas (%) de internamento, mortalidade dos internados e letalidade global em Portugal por mês. Fonte: CDC. ACálculos e análise: PÁGINA UM.
Esta situação indiciará que os internados mais vulneráveis – que necessitam de internamento – terão um risco de morte superior no caso da covid-19 do que na gripe. Mais uma vez, o tira-teimas seria uma análise estratificada, mas somente se o Tribunal Administrativo de Lisboa obrigar o Ministério da Saúde será possível retirar uma conclusão elucidativa.
Porém, ninguém, para já, pode negar uma evidência: a covid-19 de 2022 claramente não é a mesma covid-19 do passado. E mais do que as vacinas, a “chave” da mudança aparenta estar na variante Omicron, que trouxe maior transmissibilidade mas muito menor agressividade. Um sinal do seu carácter (já) endémico.
Com a espuma dos dias a desaparecer em redor da pandemia, começam a surgir investigadores com coragem para análises menos emotivas e mais científicas. Anteontem, na prestigiada BMJ Global Health foi publicado um extenso artigo de nove investigadores de diversas universidades dos Estados Unidos, Canadá e Reino Unido onde não se poupam críticas aos abusos cometidos na gestão da pandemia que colidiram “com os direitos humanos e promoveram a polarização social, afectando a saúde e o bem-estar”.
Nove investigadores norte-americanos, canadianos e britânicos acusam as políticas de vacinação contra a covid-19, seguidas pelos diversos países democráticos, de terem tido “efeitos prejudiciais na confiança do público, na confiança nas vacinas, na polarização política, nos direitos humanos, nas desigualdades e no bem-estar social”.
Num extenso artigo de 14 páginas publicado na passada quinta-feira na prestigiada revista científica BMJ Global Health, os nove investigadores – que trabalham, entre outros centros, na Universidade de Oxford, Johns Hopkins University (Maryland), London School of Hygiene & Tropical Medicine, Universidade de Washington e Universidade de Toronto – questionam “a eficácia e as consequências da política de vacinação coerciva na resposta à pandemia”, recomendando aos decisores políticos que “retomem abordagens de saúde pública não discriminatórias e baseadas na confiança.”
Intitulado “The unintended consequences of COVID- 19 vaccine policy: why mandates, passports and restrictions may cause more harm than good”, o artigo aborda, em detalhe, como foi implementada a estratégia de vacinação maciça e as suas implicações em termos de psicologia comportamental (reactância, dissonância cognitiva, estigma e desconfiança), política e direito (efeitos nas liberdades civis, polarização e governança global), socio-economia (efeitos na desigualdade, capacidade do sistema de saúde e bem-estar social) e de integridade da Ciência e da Saúde pública (a erosão da ética da saúde pública e da supervisão regulatória). E também a forma ziguezagueante como políticos e media se comportaram.
Reconhecendo que as vacinas tiveram impacto significativo na redução da taxa de mortalidade relacionada com a covid-19, os investigadores criticam sobretudo os mecanismos de coerção e estigmatização implementados nos últimos dois anos, que “provocaram considerável resistência social e política”, o que, segundo eles, tiveram “consequências prejudiciais não intencionais”, as quais “podem não ser éticas, cientificamente justificadas e eficazes.”
Primeira página do artigo.
Por exemplo, no caso da adopção dos certificados digitais, como passes sanitários para o acesso a determinados locais, os investigadores salientam que acabou por “colidir com os direitos humanos e promover a polarização social afectando a saúde e o bem-estar”, tendo sido usado com um fito “inerentemente punitivo, discriminatório e coercitivo.” Defendem, por isso, ser da máxima importância uma reavaliação “à luz das consequências negativas.”
No artigo relembra-se também a manipulação da opinião pública em redor da eficácia das vacinas ao longo do ano passado para incentivar a adesão da população.
“A lógica comunicada publicamente para a implementação de tais políticas mudou ao longo do tempo”, salientam os autores. Numa primeira fase dizia-se que a vacinação visava a “proteção dos mais vulneráveis”. Em seguida serviria para se alcançar a “imunidade de rebanho’, acabar com a pandemia’ e ‘voltar ao normal’, assim que o suprimento de vacinas fosse suficiente”. Porém,“no final do Verão de 2021” já passou a defender-se “a recomendação universal de vacinação para reduzir a pressão hospitalar e nas unidades de cuidados intensivos na Europa e América do Norte”.
Sobre as políticas gerais da vacinação obrigatória, os autores admitem que têm sido cada vez mais desafiadas e questionadas, devido à diminuição significativa da eficácia contra a infecção, apontando também que estudos realizados em Israel e no Reino Unido mostram que a “vacinação forçada aumentou os níveis de contestação, especialmente naqueles que já desconfiavam das autoridades”, agudizando a polarização social.
Neste aspecto, os media mainstream são particularmente criticados pelos investigadores, por terem usado “narrativas simplistas sobre percepções públicas complexas”, sobretudo quando sistematicamente optaram por catalogar as posições críticas como uma “consequência de forças ‘anti-ciência’ e de ‘extrema-direita”.
Nessa linha, a pressão social sobre os não-vacinados chegou a níveis de perseguição. Por exemplo, ainda que a imunidade natural – adquirida por uma infeção anterior por SARS-CoV-2, tenha fornecido uma protecção significativa, mesmo superior à da imunidade vacinal, “muitos dos que foram infetados acabaram por ser suspensos dos seus empregos ou até mesmo despedidos”, no caso de não se terem vacinado, denunciam os investigadores. “Estas pessoas, ficaram impedidas de viajar ou de participar em eventos públicos”, acrescentam.
Não ser vacinado passou a ser alvo de uma discriminação automática, incentivada por políticos e mesmo pelos media. Discriminar ou rotular não-vacinados “tornou-se socialmente aceitável entre os grupos de pró-vacinas, media e o público em geral, que viram a vacinação completa como uma obrigação moral e parte do contrato social”, referem os investigadores, mas apontam as consequências nefastas: “O efeito, no entanto, tem sido o de polarizar a sociedade – física e psicologicamente (…) A política de vacinas parece ter impulsionado as atitudes sociais em direção a uma dinâmica nós/eles em vez de adaptativa com estratégias para diferentes comunidades e grupos de risco.”
Para exemplificar, as atitudes hostis de responsáveis políticos, os investigadores elencam frases ameaçadoras e estigmatizantes de diversos políticos, como Emmanuel Macron, Justin Trudeau, Joe Biden, Jacinda Ardern e Tony Blair.
A declaração do presidente francês, feita no início de Janeiro deste ano, é bastante reveladora da procura de estigmatização: “É uma pequena minoria que está a resistir. Como reduzir essa minoria? Irritando-os ainda mais… Quando a minha liberdade ameaça a liberdade dos outros, eu passo a ser um irresponsável e alguém irresponsável não é um cidadão”.
Também a de Tony Blair é destacada: “Precisamos chegar aos não-vacinados. Francamente, se você ainda não está vacinado, se é elegível e não tem razões de saúde para não ser vacinado, você não é apenas um irresponsável, mas um idiota.” E também são salientadas duas intervenções do presidente norte-americano, uma das quais em Setembro do ano passado em que responsabilizava os não-vacinados pela manutenção da pandemia. Joe Biden garantia que se estava perante uma “pandemia de não-vacinados”. Como agora se sabe, as vacinas concedem uma protecção extremamente curta ou mesmo irrelevante na redução da transmissão.
Segundo os investigadores, “os governos abusaram [também] do poder, invocando um constante estado de emergência, evitando [assim] a consulta pública”, além de terem demonstrado “que confiavam excessivamente nos dados fornecidos pelas farmacêuticas”.
Considerando também que “a confiança nas autoridades de saúde se perde quando estas não são transparentes” – até porque não existiu transparência sobre o impacto negativo das vacinas, o que “exacerbou as ansiedades sociais, frustrações, raiva e incerteza”, os investigadores concluem que “as consequências criadas por estas circunstâncias, provocam uma tensão entre os princípios constitucionais e bioéticos, especialmente em democracias liberais”. Razão que os leva depois a relembrar que “as estruturas éticas foram projetadas para assegurar que os direitos e liberdades sejam respeitados mesmo durante a emergência de saúde pública”.
Perante a recusa sistemática de acesso a documentos administrativos por parte da Direcção-Geral da Saúde, pedidos ao longo dos últimos meses, o PÁGINA UM avançou hoje com um processo de intimação no Tribunal Administrativo de Lisboa contra o Ministério da Saúde. O processo é considerado “urgente” e já foi distribuído a uma juíza, e será a derradeira hipótese de terminar com o obscurantismo em redor da gestão política da pandemia. Conheça quais são os documentos em causa, incluindo base de dados, que a DGS tem estado a recusar ao PÁGINA UM.
A ministra da Saúde, Marta Temido, terá de se justificar perante o Tribunal Administrativo de Lisboa sobre as razões para recusar o acesso a um vasto conjunto de documentos administrativos solicitados pelo PÁGINA UM à Direcção-Geral da Saúde (DGS).
O processo de “intimação para prestação de informação e passagem de certidões” foi hoje intentado pelo director do PÁGINA UM, e como processo urgente, sob o número 1438/22.8BELSB, foi já distribuído à juíza Ilda Maria Pimenta Côco.
Apesar de ter sido a DGS a recusar sistematicamente o fornecimento de documentos administrativos, incluindo o acesso a base de dados, do ponto de vista formal o réu, neste processo, será o Ministério da Saúde.
Marta Temido, ministra da Saúde, tem acompanhado a pandemia da covid-19 desde o início.
A decisão do PÁGINA UM decorre de longas e pacientes tentativas de obtenção de documentação relacionada com o sistema de informação e de gestão da pandemia da covid-19, cujos pedidos têm sido quase todos recusados pela directora-geral da Saúde Graça Freitas.
Apesar de diversos pareceres não-vinculativos já emitidos pela Comissão de Acesso aos Documentos Administrativos (CADA), instando a DGS a fornecer o acesso a um vasto conjunto de documentos essenciais para a compreensão da dimensão e amplitude da pandemia, e das respostas políticas, a DGS somente por uma vez disponibilizou dados: os pareceres da Comissão Técnica de Vacinação contra a Covid-19. Porém, recusou disponibilizar as actas das reuniões, de modo a esconder o sentido de voto dos membros que, por exemplo, se opuseram à estratégia de vacinação dos adolescentes.
Com a intimação agora apresentada, o Tribunal Administrativo de Lisboa poderá, no prazo de sensivelmente um mês, decidir pela obrigatoriedade no fornecimento dos documentos administrativos. E, neste caso, o próprio Ministério da Saúde vai ser mesmo obrigado a justificar os motivos de manter um secretismo absoluto sobre documentos administrativos relacionados com a covid-19.
Este processo de intimação insere-se na campanha do PÁGINA UM em prol da defesa da informação científica e da transparência, sendo integralmente financiada pelo FUNDO JURÍDICO, através de donativos dos leitores na plataforma MIGHTYCAUSE, tendo como patrono o advogado Rui Amores, especialista em Direito Administrativo. Este é o sexto processo intentado pelo PÁGINA UM.
Conheça aqui quais são os documentos solicitados pelo PÁGINA UM ao Ministério da Saúde como entidade que tutela a DGS:
1 – Actas de todas as reuniões da Comissão Técnica de Vacinação contra a Covid-19, criada pelo despacho de V. Exa. com o número 012/2020 de 4 de Novembro de 2020.
Graça Freitas, directora-geral da Saúde, tem sistematicamente recusado responder aos pedidos do PÁGINA UM. Tudo pode mudar com a intervenção do Tribunal Administrativo.
2 – Base de dados do Sistema Nacional de Vigilância Epidemiológica (SINAVE), a plataforma que tem vindo a ser usada para acompanhamento da pandemia causada pelo vírus SARS-CoV-2, devendo ser autorizado o acesso presencial à referida base de dados, e onde conste a seguinte informação detalhada (para cada um dos casos positivos reportados pelos médicos e laboratórios): a) Data da confirmação do teste positivo b) Identificação da pessoa (com id anonimizado) c) Idade à data da validação d) Nacionalidade do utente e) Concelho do utente f) Variante do vírus (se identificada) g) Situação da vacinação (vacinada parcialmente com uma dose; vacinação completa; vacinação completa com dose de reforço; não-vacinada) h) Marca da vacina (se vacinado) i) Data do óbito (se ocorreu).
3 – Dados anonimizados de todos os óbitos registados no Sistema de Informação dos Certificados de Óbito (SICO) desde 2013 até à data, onde conste (obviamente sem identificação da pessoa) a data do óbito, a idade da pessoa em causa, o local do óbito (concelho) e a causa apurada do óbito de acordo com o código respectivo da Classificação Estatística Internacional de Doenças e Problemas Relacionados com a Saúde (CID), devendo assim ser expurgados os dados que possam identificar, mesmo que indirectamente, a pessoa em causa. Se se considerar que a indicação do local do óbito (concelho) seja susceptível de identificar qualquer pessoa, então que se opte pela identificação do local por distrito. E se se considerar que até com o distrito seja passível de uma identificação, então prescinde-se da identificação do local do óbito, desde que os outros elementos solicitados estejam presentes. Pode, e deve, ser expurgado o nome do médico legista.
4 – Documentos administrativos que contenham o registo do número de testes de detecção de SARS-CoV-2 por idade (desagregada por idade ou agregada por faixa etária) em cada dia, desde o início da pandemia, quer sejam testes PCR quer testes de antigénio, bem como os documentos administrativos que contenham o registo do número de casos positivos por idade (desagregada por idade ou agregada por faixa etária) em cada dia, desde o início da pandemia, quer sejam testes PCR quer testes de antigénio.
5 – Documentos administrativos que contenham o registo (ou cujos dados permitam apurar) sobre a evolução (temporal) da incidência cumulativa (real ou estimada) e as taxas de letalidade em Portugal das diferentes variantes classificadas pela Organização Mundial da Saúde (OMS) como de preocupação (VOC), designadamente a Alpha, Beta, Gamma, Delta e Omicron, e de interesse (VOI), designadamente a Lambda e Mu.
6 – Documentos administrativos que contenham o registo do número de surtos de covid-19 em unidades hospitalares – isto é, que a covid-19 seja considerada infecção nosocomial –, discriminados por unidade e mês (ou outro qualquer período temporal), integradas no Serviço Nacional de Saúde (SNS), desde o início da pandemia até à data da consulta a efectuar.
7 – Documentos administrativos que contenham o registo com o número total de infecções (casos positivos) por covid-19, e eventualmente discriminadas por unidade hospitalar e por mês (ou outro qualquer período temporal), adquiridas durante o internamento por outras causas, ou seja, que seja possível aferir do número de infecções nosocomiais de covid-19, desde o início da pandemia até à data da consulta a efectuar.
8 – Documentos administrativos que contenham o registo com o número total de óbitos atribuídos à covid-19 em doentes previamente internados por causas não-covid e que sofreram infecção nosocomial de covid-19 durante o internamento, e eventualmente discriminados por unidade hospitalar e por mês (ou outro qualquer período), desde o início da pandemia até à data da consulta a efectuar.
9 – Documentos administrativos que contenham informação detalhada, desde o início da pandemia, até ao momento da consulta, relacionada com o internamento de doentes com teste positivo à covid-19 (internados-covid). Basicamente, aquilo que se solicita é a base de dados, convenientemente anonimizada, que a DGS confirmou em 4 de Fevereiro p.p. a sua existência, através de comunicado de imprensa, onde se destaca que cerca de 75% das pessoas consideradas doentes-covid estiveram internadas por consequência direta dessa infeção.
10 – Documentos administrativos que contenham informação desde o início da pandemia, até ao momento da consulta, sobre o número de utentes, por Estrutura Residencial para Pessoas Idosas (ERPI), cujos óbitos tenham ocorrido numa instituição com casos confirmados de covid-19 ou em utente ou trabalhador que tenha apresentado sintomas compatíveis com a doença. Em suma, pretende-se ter acesso, consultar e obter cópia integral de todas as comunicações recebidas pela DGS, ou o suporte digital dessas comunicações após tratamento informático, em cumprimento do ponto 68 da Orientação nº 009/2020 de 11 de Março de 2020, com actualização em 10 de Janeiro p.p.. Ou, em alternativa, um documento oficial já existente que contenha, de forma clara, e discriminada, essa informação
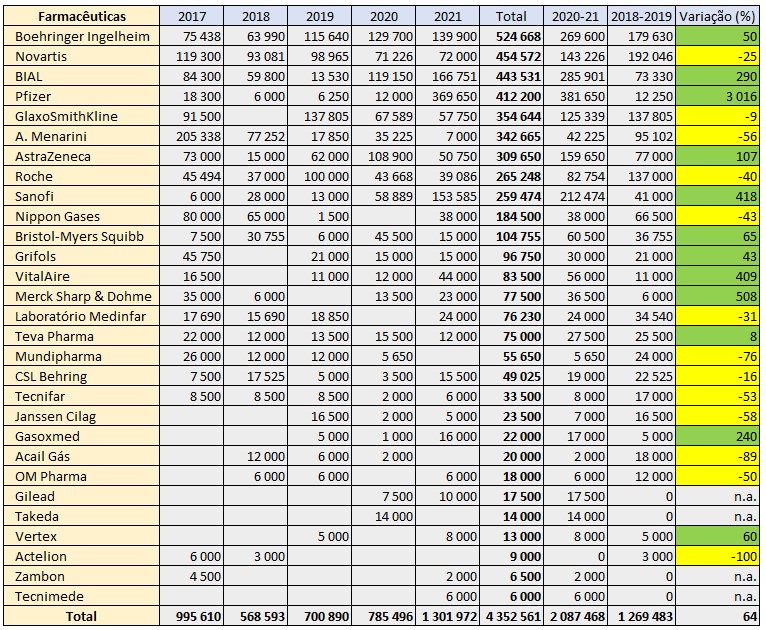
António Morais acumula a presidência da Sociedade Portuguesa de Pneumologia (SPP) com as funções de consultor da Direcção-Geral da Saúde e do Infarmed. Lei diz que só poderia acumular se a SPP recebesse das farmacêuticas no máximo 50.000 euros por ano em média no quinquénio anterior. A SPP recebeu no período 2017-2021 cerca de 870 mil euros, ou seja, 17 vezes mais.
A Inspecção-Geral das Actividades em Saúde (IGAS) está a investigar o presidente da Sociedade Portuguesa de Pneumologia, António Morais. A abertura formal de um “Processo de Esclarecimento, o qual se encontra em curso” foi admitida pelo inspector-geral desta entidade, António Carapeto, em carta a que o PÁGINA UM teve acesso.
De acordo com o IGAS, um processo de esclarecimento deste tipo constitui um “procedimento rápido e expedito destinado à recolha de elementos com vista ao esclarecimento de expediente geral, à verificação prévia de requisitos que habilitem a eventual decisão de instauração de acção inspectiva ou ao acompanhamento de acções inspectivas em curso dentro ou fora” desta entidade.
António Morais (ao centro), preside à Sociedade Portuguesa de Pneumologia, e é consultor da DGS e do Infarmed.
Na base da abertura desta investigação está a notícia do PÁGINA UM de 18 de Abril passado que denunciou que António Morais está a violar há três anos, desde que tomou posse como presidente da SPP, as regras de incompatibilidade que o deveriam impedir de se manter como consultor do Infarmed e da Direcção-Geral da Saúde. As decisões administrativas que tenham sido tomadas com base em pareceres em que este pneumologista tenha participado são juridicamente nulas.
António Morais – que desde 2016, e apresenta-se como tal no seu currículo, é consultor de doenças intersticiais pulmonares do Programa Nacional para as Doenças Respiratórias da Direcção-Geral da Saúde (DGS) e membro da Comissão de Avaliação de Tecnologias de Saúde do Infarmed – não poderia estar a acumular aquelas funções públicas com as de membro dos órgãos sociais de uma sociedade profissional com tão estreitas relações comerciais com farmacêuticas.
Um decreto-lei de 2014 estipula que consultores, membros de comissões, grupos de trabalho e júris de concursos com determinadas funções em organismos do Ministério da Saúde não podem ser, em simultâneo, membros de órgãos sociais de sociedades científicas – como é o caso da SPP – que “tenham recebido financiamentos de empresas produtoras, distribuidoras ou vendedoras de medicamentos ou dispositivos médicos, em média por cada ano num período de tempo considerado até cinco anos anteriores, num valor total superior a 50.000”.
Ora, António Morais preside à SPP desde 14 de Janeiro de 2019, e esta sociedade médica ultrapassa larguissimamente o patamar dos 50 mil euros anuais. Quando este pneumologista – que exerce no Hospital de São João e na Trofa Saúde, além de ser também professor na Faculdade de Medicina do Porto – tomou posse, a SPP tinha recebido no quinquénio anterior uma média de 799.634 euros do sector farmacêutico, ou seja, 16 vezes mais do que o limite imposto pela norma das incompatibilidades.
No quinquénio 2017-2021, que engloba já os três anos de presidência de António Morais, os montantes arrecadados pela SPP ainda aumentaram mais: situaram-se nos 870.512 euros por ano. Para este aumento muito contribuiu o ano passado em que a SPP recebeu um financiamento recorde vindo do sector farmacêutico: 1.301.972 euros.
Em 2022, até ao dia de hoje, de acordo com a Plataforma da Publicidade e Transparência do Infarmed, a SPP amealhou 499.228 euros, mas usualmente a maior fatia de patrocínios e contratos comerciais com a indústria farmacêutica regista-se no último trimestre de cada ano no âmbito do Congresso de Pneumologia.
A título pessoal, António Morais tem também relações comerciais com farmacêuticas. Este ano já recebeu 10.281 euros provenientes de sete farmacêuticas.
Apoios do sector farmacêutico (em euros) à Sociedade Portuguesa de Pneumologia entre 2017 e 2021. Fonte: Infarmed.
Para além de questões éticas, as incompatibilidades de António Morais têm consequências legais e jurídicas muito graves. De acordo com o artigo 5º do Decreto-Lei nº 14/2014, “os pareceres emitidos ou as decisões tomadas por comissões, grupos de trabalho, júris e consultores, em que intervenham elementos em situação de incompatibilidade não produzem quaisquer efeitos jurídicos”, o que significa, em consequência, que “as decisões dos órgãos deliberativos (…) são nulas”, caso se baseiem naqueles pareceres.
António Morais, por seu turno, pode vir também a ser sancionado, porque o artigo 6º do mesmo diploma legal determina a obrigatoriedade de ele cessar as suas funções de consultor a partir do dia de tomada de posse como presidente da SPP (14 de Janeiro de 2019). O PÁGINA UM teve acesso à sua última declaração, com data de 5 de Março de 2018 – numa altura, portanto, em que ainda não presidia à SPP, e não estaria a violar o regime de incompatibilidades –, e que ainda consta no site do Infarmed.
Por essa falha, a IGAS pode, de acordo com a lei, aplicar-lhe uma coima entre 2.000 e 3.500 euros. Ou, simplesmente, não fazer nada, e a SPP continuar a receber aqueles montantes das farmacêuticas, tendo um presidente a aconselhar a DGS e o Infarmed como se fosse um perito independente.
O PÁGINA UM analisou o desempenho do Serviço Nacional de Saúde (SNS) ao longo da pandemia, entre Março de 2020 e Janeiro de 2022, face aos períodos anteriores. Duas evidências: Janeiro de 2021 foi um descalabro inimaginável nos hospitais portugueses; e a culpa não foi apenas da covid-19. Houve “departamentos” hospitalares importantes que pioraram as taxas de mortalidade ao longo da pandemia, mesmo com muito menos doentes.
O colapso do Serviço Nacional de Saúde durante a pandemia, sobretudo no Inverno de 2020-2021 – em que se assistiu a um recorde de mortes nos hospitais portugueses –, não se deveu somente aos casos de covid-19.
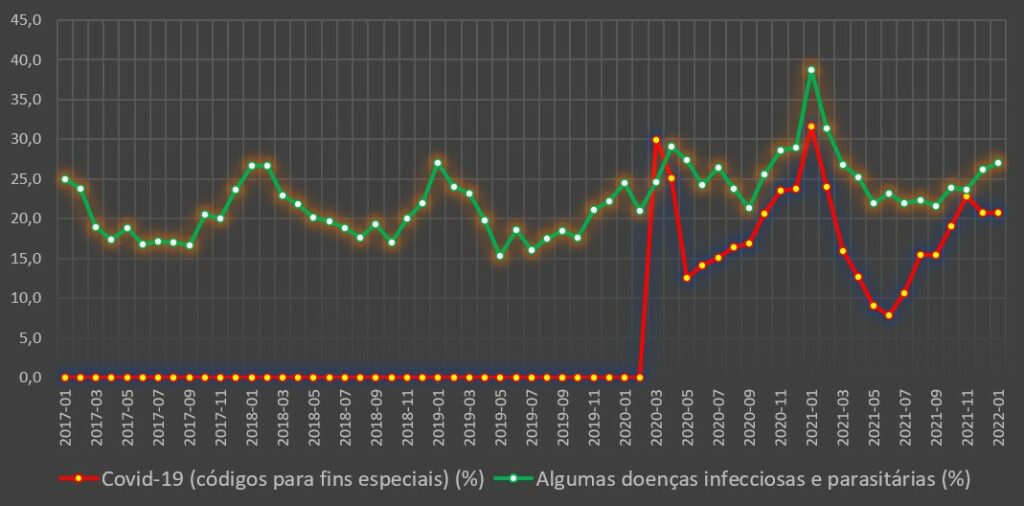
Mais uma análise do PÁGINA UM à base de dados da morbilidade e mortalidade do Portal da Transparência do SNS revela, desta vez, que o incremento na mortalidade hospitalar, em especial em Janeiro de 2021, atingiu níveis elevados sobre os internados com covid-19. Mas também os internados por doenças do aparelho respiratório e por doenças infecciosas e parasitárias (códigos A e B da CID – Classificação Internacional de Doenças), e outras doenças, tiveram menores chances de sobrevivência do que aqueles que sofreram dos mesmos males antes da pandemia.
De acordo com os registos dos internamentos e dos óbitos por mês, desde 2017, para cada grupo de doenças, o mês de Janeiro do ano passado mostrou uma situação catastrófica nos hospitais portugueses, com uma taxa de mortalidade global de 14,1%. Em termos comparativos, o mês homólogo nos quatro anos anteriores situou-se entre 6,9% em 2020 e 7,6% em 2017. Este ano, este rácio “normalizou”, fixando-se em 7,5%.
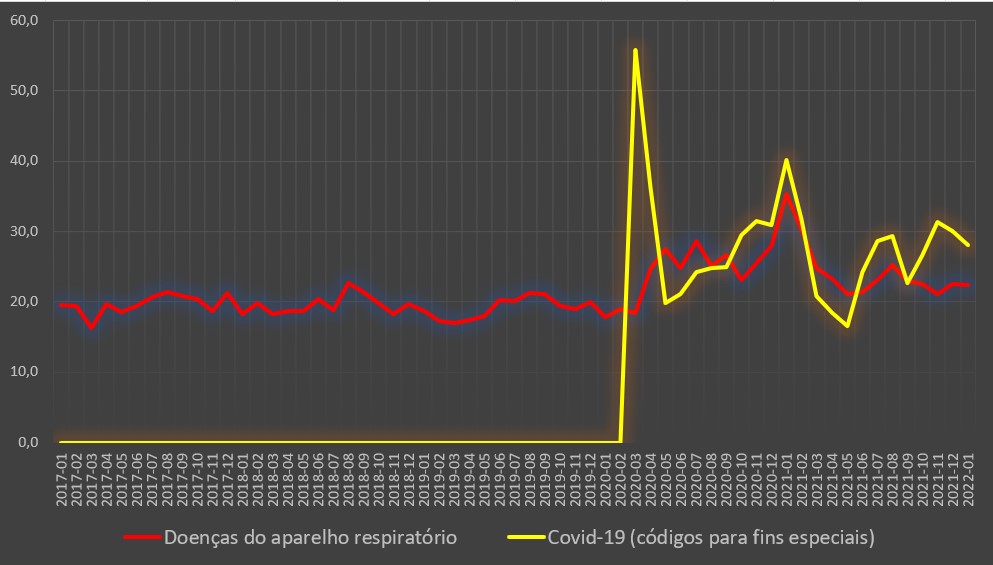
O peso da covid-19 para este descalabro foi importante, mas longe de ser único. Com efeito, em Janeiro de 2021, efectivamente a mortalidade hospitalar dos internados atingiu valores máximos (31,7%), muito acima do valor médio desta doença desde que surgiu em Portugal a partir de Março de 2020 (22,4%).
Variação da taxa de mortalidade hospitalar (%) por mês para a covid-19 e para todas as doenças. Fonte: SNS. Cálculos e análise: PÁGINA UM.
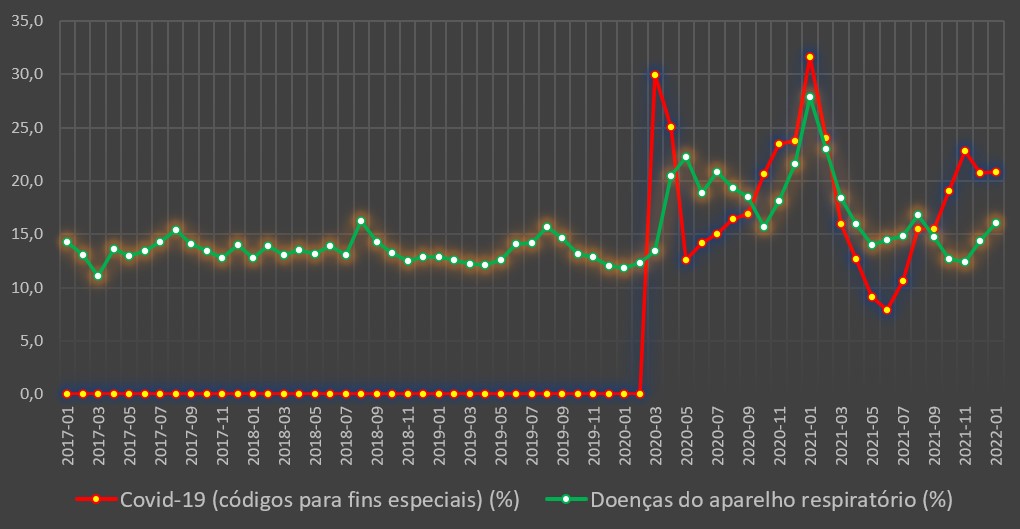
Porém, sobretudo nas doenças infecciosas e parasitárias, e também nas doenças do aparelho respiratório, o mês de Janeiro de 2021 foi também de hecatombe. Ou seja, quem esteve internado com doenças daqueles tipos nos hospitais viu a sua chance de sobrevivência baixar significativamente.
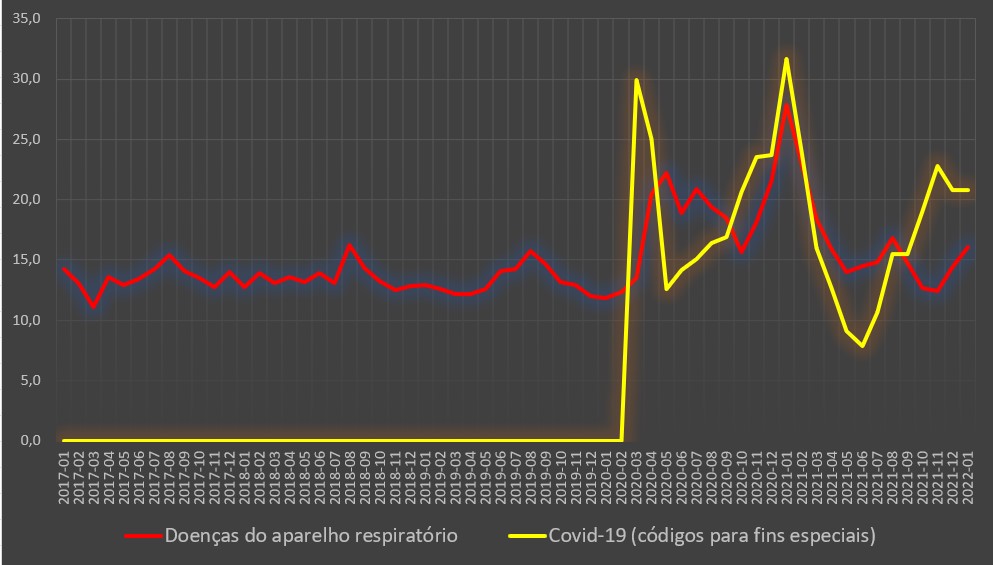
No caso dos internados por doenças do aparelho respiratório, a taxa de mortalidade em Janeiro de 2021 foi de 27,8%, muito mais do dobro dos valores registados no mês homólogo dos quatros anos anteriores.
De facto, no ano imediatamente anterior – em vésperas da chegada da covid-19 e num período em que a gripe e subsequentes infecções respiratórias estavam pouco agressivas –, a taxa de mortalidade hospitalar situou-se apenas nos 11,8%. Nos anos anteriores foi um pouco mais elevada, mas longe do desastre de 2021: atingiu os 14,2% em 2017 (com um surto gripal de alguma agressividade), e foi de 12,7% e 12,9% em 2018 e 2019, respectivamente.
Variação da taxa de mortalidade hospitalar (%) por mês para a covid-19 e para as doenças do aparelho respiratório. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Este ano, a taxa de mortalidade hospitalar por doenças respiratórias foi de 16,1%, muito inferior ao valor do ano passado, mas mesmo assim bastante superior aos valores normais para esta época do ano.
Relativamente às doenças infecciosas e parasitárias dos grupos A e B do CID, a situação em Janeiro de 2021 foi também dramática, tendo a taxa de mortalidade hospitalar atingido os 38,7%, ou seja, mesmo acima da covid-19 para aquele mês. No mês homólogo de 2017 a 2020, esta taxa situou-se no intervalo entre 24,5% e 27,0%. Em Janeiro deste ano, este rácio já se normalizou, tendo ficado nos 27,1%.
Embora o mês de Janeiro de 2021 evidencie um agravamento colossal – na verdade, um colapso – da capacidade de resposta do SNS, apesar da redução de 270 mil internados em 2020 e 2021 face ao biénio anterior, ao longo da pandemia as taxas de mortalidade pioraram em quase todos os grupos de doenças.
Variação da taxa de mortalidade hospitalar (%) por mês para a covid-19 e para as doenças infecciosas e parasitárias (códigos A e B). Fonte: SNS. Cálculos e análise: PÁGINA UM.
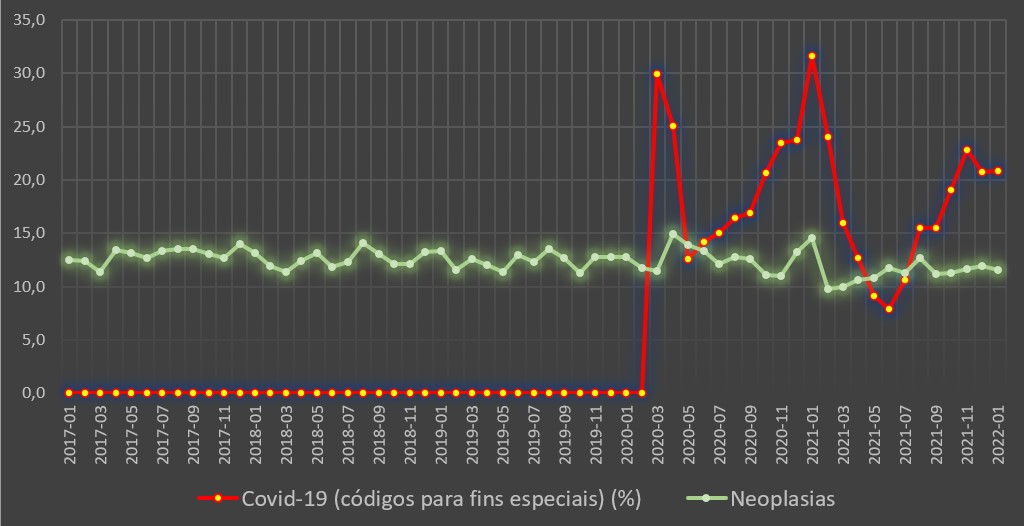
Com efeito, de entre os grupos de doenças de prognóstico de internamento mais incerto – com taxa de mortalidade hospitalar acima dos 10% antes da pandemia –, apenas nas neoplasias se observou uma ligeira redução, passando de 12,5% nos 23 meses anteriores à pandemia (Abril de 2018 a Fevereiro de 2020) para os 11,9% entre Março de 2020 e Janeiro de 2022 (23 meses).
Contudo, durante a pandemia, face ao período anterior, foram internadas menos 33.175 pessoas com doenças oncológicas. Ou, pelo menos, não foram internadas como sofrendo de cancros. Nem os óbitos, se ocorreram, tiveram essa causa apontada.
Variação da taxa de mortalidade hospitalar (%) por mês para a covid-19 e para as neoplasias. Fonte: SNS. Cálculos e análise: PÁGINA UM.
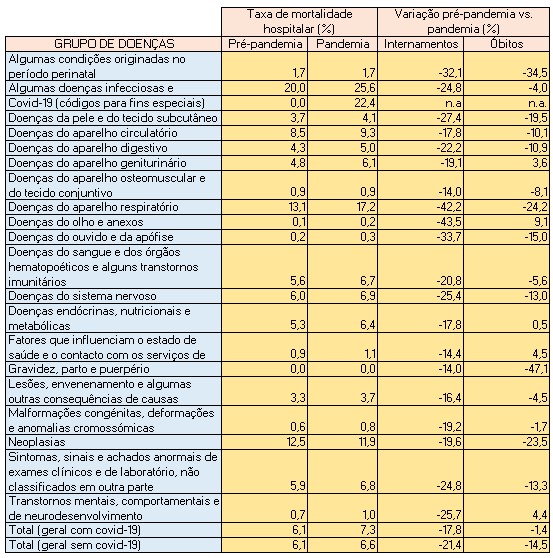
Mas nos casos das doenças infecciosas e parasitárias dos grupos A e B e das doenças do aparelho respiratório a taxa de mortalidade média durante a pandemia foi substancialmente superior à do período anterior. No primeiro grupo subiu de 20,0% para 25,6%; no segundo grupo cresceu de 13,1% para 17,2%.
Em termos globais, incluindo a covid-19, e confrontando os dois períodos acima referidos, a taxa de mortalidade hospitalar subiu de 6,1% para 7,3%. Significa que a taxa de mortalidade hospitalar sofreu um agravamento de 20%. Porém, se se retirar os internamentos e óbitos da covid-19, o agravamento para as outras doenças também se verifica, embora em menor grau (mais 9%), passando de 6,1% para 6,6%.
Taxas de mortalidade hospitalar por grupo de doenças no período pré-pandémico (Abril de 2018 a Fevereiro de 2020) e pandémico (Março de 2020 a Janeiro de 2022) e variações de internados e de óbitos. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Convém, contudo, destacar que, se se não contabilizar os internados-covid, os internamentos por todas as outras doenças entre Março de 2020 e Janeiro de 2022 decaíram 21,4% (menos 360.266 internamentos) face ao período entre Abril de 2018 e Fevereiro de 2020.
Essa variação deve-se sobretudo à queda nos internamentos das doençasdo aparelho respiratório, em parte devido ao “desaparecimento”da gripe (e das pneumonias associadas) durante a pandemia.
Mas assistimos asima um estranho paradoxo: uma menor pressão hospitalar nas áreas dedicadas a doenças não-covid acabou por resultar, afinal, num agravamento das respectivas taxas de mortalidade, o que mostra que nem todas as responsabilidades sobre o excesso de mortalidade se pode assacar ao SARS-CoV-2 e à covid-19.
Existe, contudo, um aspecto que deverá merecer maior investigação.
O agravamento das taxas de mortalidade nas outras doenças não se deveu a um maior número de óbitos – na maior parte dos grupos de doenças houve um decréscimo absoluto –, o que pode indiciar que tanto os internamentos como os óbitos em determinadas doenças estarão subestimados porque foram “endossados” à covid-19 apenas devido a, no momento da hospitalização, os doentes estavam com teste positivo.
Nota: Nesta análise, as taxas de mortalidade foram calculadas em função do número de óbitos e de internamentos ocorridos em cada mês. Obviamente, este indicador mensal não reflecte a taxa efectiva de mortalidade durante cada um dos períodos (ou, se assim se desejar, o risco de morte por internamento), porque os óbitos ocorridos em determinado mês são também de doentes internados em meses anteriores. No entanto, este rácio, assim calculado, e na falta de dados mais discriminados, constitui um adequado indicador de desempenho do SNS.
Uma análise de dados oficiais feita pelo PÁGINA UM revela que dar prioridade máxima ao tratamento da covid-19 teve um efeito secundário inesperado (ou não): os internados por doenças respiratórias não-covid tiveram um risco acrescido de morte. E a grande surpresa é que, em determinados períodos, sobretudo na Primavera e Verão de 2020 e 2021, as doenças respiratórias até registaram taxas de mortalidade hospitalar superiores à da covid-19. E mais: a opção inicial de entubar doentes idosos terá sido catastrófica.
Durante a pandemia, entre Março de 2020 e Janeiro de 2022, a taxa de mortalidade hospitalar dos doentes-covid foi apenas 30% superior à registada nos internados com doenças respiratórias. Esta é uma das principais revelações da análise feita pelo PÁGINA UM aos dados da morbilidade e mortalidade do Portal da Transparência do Serviço Nacional de Saúde (SNS). Mas esta situação deveu-se também a um outro problema: com a pandemia, e uma priorização à covid-19, os doentes com doenças respiratórias não-covid viram a sua chance de sobrevivência diminuir.
De acordo com a análise, a taxa de mortalidade hospitalar dos internados-covid – medida de uma forma simplista, face à ausência de informação mais detalhada, pelo número de mortes em cada mês em função dos internados nesse mês – foi de 22,4% entre Março de 2020 e Janeiro deste ano. Ou seja, em cada 1.000 internados acabaram por morrer 224.
Essa taxa é calculada face ao número oficial de internamentos por covid-19 nos hospitais públicos naquele período (59.916 pessoas) e ao número efectivo de óbitos nos hospitais do SNS causados por covid-19 (13.397 mortes).
Convém referir que o Ministério da Saúde não explicou ainda como cerca de um terço dos óbitos por covid-19 anunciados pela Direcção-Geral da Saúde não ocorreram afinal numa unidade de saúde, face à infecciosidade da doença e ao facto de o agravamento do estado de saúde recomendar sempre um internamento.
Em todo o caso, esta taxa de mortalidade hospitalar da covid-19 (22,4%) pode ser considerada bastante mais elevada face ao que se registava no período pré-pandemia para as outras doenças respiratórias, mas já não tanto naquilo que veio a suceder durante o período pandémico.
Com efeito, segundo os dados do SNS, entre Janeiro de 2017 e Fevereiro de 2020 (38 meses), a taxa de mortalidade hospitalar em internados por doenças respiratórias foi de 13,2%, correspondente a 43.715 óbitos em 330.341 internados.
No entanto, com o surgimento da pandemia – e a menor atenção concedida a todas as outras afecções –, a taxa de mortalidade hospitalar por doenças respiratórias deu um pulo, atingindo um agravamento de 4 pontos percentuais.
Ou seja, se antes da pandemia, por cada 1.000 internados por doenças respiratórias morriam 132 pessoas, após Março de 2020 passaram a morrer 173 por cada mil. Este agravamento também se observa pela variabilidade da taxa de mortalidade.
Taxa de mortalidade (%) geral dos internados nos hospitais do SNS por mês desde Janeiro de 2017 por doenças do aparelho respiratório e por covid-19. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Se antes da pandemia, o risco de mortes nos hospitais por doenças respiratórias não sofria grandes variações ao longo do ano – variando entre os 11% e os 16% –, os “desarranjos” nos hospitais do SNS causaram oscilações caóticas, superando em alguns meses os 20%.
Em determinados períodos, a taxa de letalidade das doenças respiratórias chegou a ser mesmo superior à da covid-19 em dois períodos longos: entre Março e Setembro de 2020 e entre Março e Agosto de 2021.
Mesmo no pico da letalidade da covid-19 – Janeiro de 2021 –, em que a taxa de mortalidade desta doença atingiu um máximo de 31,7% (ou seja, quase uma em cada três pessoas internadas por causa do SARS-CoV-2 acabaram por não sobreviver), a taxa de mortalidade hospitalar por doenças respiratórias alcançou os 24%, isto é, o dobro da situação habitual num Inverno.
Mas a análise do PÁGINA UM também conseguiu destacar os níveis diferentes de letalidade em função da idade dos internados, confirmando não apenas que o risco é incomensuravelmente superior nos mais idosos, mas também indiciando que, na fase inicial da pandemia, algo terá corrido mesmo muito mal nas decisões terapêuticas, sobretudo nos maiores de 65 anos.
De facto, se se confrontar a taxa de mortalidade dos menores de 65 anos, a covid-19 não se mostrou uma catástrofe em termos efectivos nesta faixa etária: em cada 1.000 internados, 58 não sobreviviam. Se se analisar os mais jovens, então o risco de morte foi extremamente baixo.
Taxa de mortalidade (%) dos internados com menos de 65 anos nos hospitais do SNS por mês desde Janeiro de 2017 por doenças do aparelho respiratório e por covid-19. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Contudo, mesmo assim também a gestão hospitalar no período pandémico permitiu que as doenças respiratórias neste grupo etário se agravassem. Se antes da pandemia, raramente a taxa de mortalidade hospitalar por doenças respiratórias nos menores de 65 anos se situava acima dos 3%, com o surgimento do SARS-CoV-2 o panorama mudou.
Em alguns meses, as doenças respiratórias não-covid registaram uma taxa de letalidade nesta faixa etária acima dos 5%, atingindo mesmo os 8,3% em Janeiro de 2021. Releve-se, contudo, que naquele mês a covid-19 atingiu um pico de 9,9% de mortalidade nos internados nesta faixa etária, mas esse foi um período de completo colapso do SNS.
Quanto ao risco de morte por covid-19 nos internados com mais de 65 anos, a análise do PÁGINA UM apurou que foi mais de cinco vezes superior (12.178 óbitos em 38.797 internados, ou seja, 31,4%) ao da faixa etária dos menores de 65 anos. Neste caso, se se comparar com a letalidade das doenças respiratórias, a covid-19 teve, sem dúvida um impacte significativo, mas longe de constituir uma catástrofe inédita.
Com efeito, no período de Janeiro de 2017 a Fevereiro de 2020, a taxa de mortalidade destas doenças rondavam os 192 óbitos por 1.000 internamentos. Significa, assim, que a covid-19 constituiu um acréscimo de risco de morte 64% face às doenças do aparelho respiratório para o grupo dos mais vulneráveis.
Taxa de mortalidade (%) dos internados com mais de 65 anos nos hospitais do SNS por mês desde Janeiro de 2017 por doenças do aparelho respiratório e por covid-19. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Porém, a pandemia trouxe também, como atrás referido, um agravamento significativo do risco de morte pelas habituais doenças respiratórias, uma vez que a taxa de mortalidade hospitalar subiu, nesta faixa etária, para 24,5%, quando antes da pandemia se situava nos 19,2%.
Relevante também é observar que a taxa de mortalidade atingiu valores perfeitamente absurdos em dois períodos para os maiores de 65 anos: em Janeiro de 2021 (com uma taxa de 40,1%) e em Março de 2020 (55,9%). No primeiro caso, deveu-se, em grande medida ao enorme fluxo de internamentos, a par de uma vaga de frio e do colapso do SNS.
Já quanto a Março de 2020 – o primeiro mês da pandemia em Portugal –, a elevada taxa de mortalidade hospitalar terá sido devido à opção, então seguida em outros países, como a Itália, de colocar todos os doentes com dificuldades respiratórias, mesmo idosos, em ventilação mecânica. A prática médica viria a revelar que esta foi uma opção com graves efeitos negativos.
Nota: Saliente-se que a taxa de mortalidade hospitalar não deve ser confundida com a taxa de letalidade de uma doença, que se mede em função dos óbitos por caso positivo, e independentemente do grau de gravidade. Não deve ser também confundido com a taxa de internamento. Destaque-se que até Janeiro deste ano se registaram cerca de 2,7 milhões de casos positivos, pelo que, tendo havido 59.916 internamentos, se contabiliza apenas uma taxa de internamento de 2,2%. Ou seja, por cada 1.000 casos positivos, 22 são internados. Se 22,4% dos internados acabam por não sobreviver, a taxa de letalidade é, deste modo, de 0,5%. Ou seja, 5 óbitos por cada 1.000 casos positivos.
A gestão da pandemia, com a criação dos “covidários” e o adiamento de muitas intervenções cirúrgicas não aliviou apenas os hospitais; fez “desaparecer” hospitalizações em todas as unidades de tratamento de doenças. Se nas alas covid e nas unidades de cuidados intensivos se deu o ‘litro’, em muitos outros departamentos houve médicos e outros profissionais de saúde que tiveram vida folgada durante a pandemia. Um paradoxo, porque em 2020 e 2021 se registou um acréscimo de mortalidade de 23 mil óbitos em Portugal, dos quase 19 mil atribuída à covid-19, embora para estes casos aplicando-se critérios muito discutíveis.
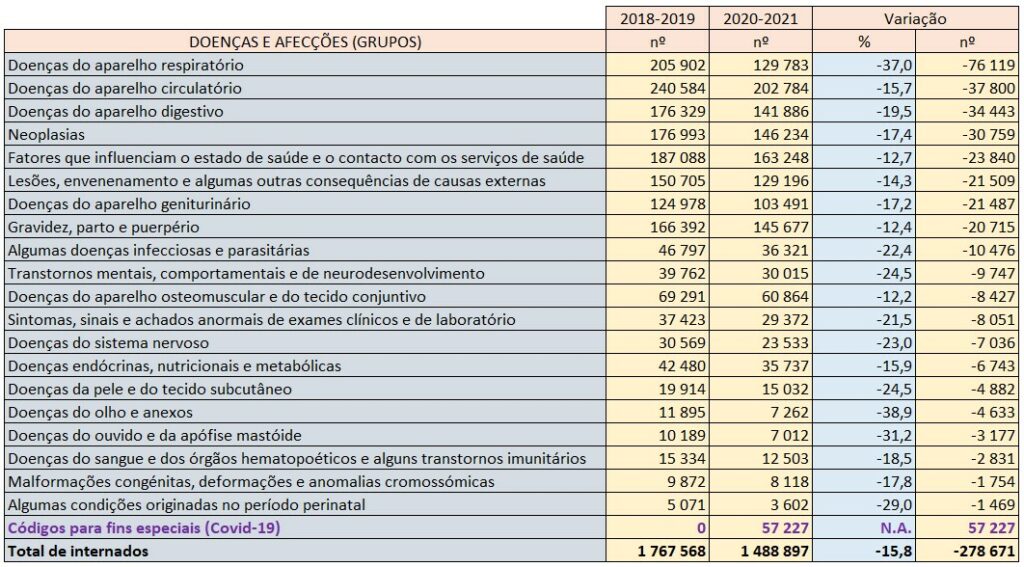
A covid-19 causou uma paradoxal redução generalizada dos internamentos em todas as valências hospitalares. De acordo com a análise do PÁGINA UM à base de dados da morbilidade e mortalidade do Serviço Nacional de Saúde (SNS), durante 2020 e 2021 – os dois primeiros anos da pandemia – registaram-se quase menos 280 mil pessoas internadas do que nos dois anos anteriores (2018 e 2019).
Isto mesmo considerando que a covid-19 – a única doença que integra o grupo de “códigos para fins especiais” –, que só surgiu no final do primeiro trimestre de 2020 contribuiu com 57.227 internados entre Fevereiro de 2020 e Dezembro de 2021.
Um dos aspectos mais surpreendentes destes dados, agora analisados pelo PÁGINA UM, é a forte queda de internamentos por todas as causas, e envolvendo mesmo áreas sem qualquer ligação directa à covid-19.
Em certa medida, esta redução deveu-se à criação dos “covidários”, para onde seguiam, independentemente da gravidade, todas as pessoas a necessitarem de cuidados médicos, mesmo se sofressem de outros problemas de saúde mais prementes.
Contudo, também se deveu muito à redução das intervenções cirúrgicas com internamento – que resultaram de uma estratégia política – e, de igual modo, ao medo incutido que afastou muitas pessoas de irem aos hospitais mesmo em caso de sintomas agudos de elevada gravidade. Muitos terão morrido por esta opção. Recorde-se que se registou um acréscimo de mortalidade no biénio 2020-2021, face a 2018-2019, de 23.017 óbitos, sendo que 18.974 foram atribuídos à covid-19.
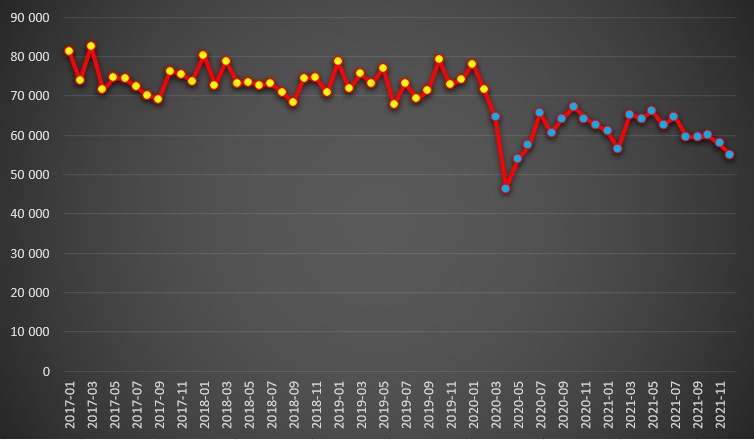
A queda no número de internados por todas as causas observou-se de forma marcante logo em Março de 2020. Com efeito, nos três anos anteriores à pandemia, os hospitais do SNS recebiam habitualmente entre 70 mil e 80 mil pessoas a necessitarem de internamento em cada mês, mas no início da pandemia, em Março de 2021, baixou para um pouco menos de 65 mil. Curiosamente, os dados do SNS indicam que houve um doente internado com covid-19 ainda em Fevereiro de 2020.
Em Abril de 2020 ainda desceu mais: 46.558 pessoas foram hospitalizadas. Nos meses seguintes, e até Dezembro do ano passado, o número de pessoas hospitalizadas por mês nunca recuperaram para os níveis pré-pandémicos.
Durante a pandemia, o mês com mais internados por todas as causas foi Outubro de 2020 com 67.080 pessoas. Em Dezembro do ano passado foram hospitalizadas apenas de 55.070 pessoas, um valor atípico. Por exemplo, no último Dezembro antes da chegada da pandemia tinham sido internadas 74.087 pessoas.
Número de pessoas internadas por mês (entre Janeiro de 2017 e Dezembro de 2021) por todas as causas em hospitais públicos. Fonte: SNS. Cálculos e análise: PÁGINA UM.
As unidades de tratamento hospitalar das doenças do aparelho respiratório não-covid foram as que mais “beneficiaram” com o surgimento da pandemia, sem prejuízo da covid-19 exigir uma logística e tratamento mais complexo. No entanto, tendo em conta que o SARS-CoV-2, a par com as medidas não-farmacológicas – uma redução substancial (ou desaparecimento efectivo) de vírus e bactérias causadoras de doenças respiratórias, os hospitais acabaram por beneficiar, nesse aspecto, de uma redução significativa da procura para tratamento.
Com efeito, de acordo com os dados do SNS, no biénio 2020-2021 foram internadas por doenças respiratórias não-covid menos 76.119 pessoas do que em 2018-2019. Significa isto que se se juntar os internados por covid-19 em 2019 e 2020 (um total de 57.227) aos internados por doenças respiratórias não-covid nesse período, então conclui-se que em 2018-2019 as unidades de pneumologia do SNS tiveram um fluxo maior de doentes.
Número de internados por mês (desde Janeiro de 2017 a Dezembro de 2021) de doenças do aparelho respiratório e de covid-19. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Na verdade, embora com taxa de letalidade maior do que a das pneumonias vulgares para as populações mais idosas, a covid-19 não implicou uma pressão descomunalmente superior nos hospitais do SNS, uma vez que se registou uma profunda queda no número de internados por pneumonias e doenças afins.
Se no período de 2017-2019 o número de internados por mês devido a doenças respiratórias se situava entre os 5.000 e os 15.000 – com os valores mais baixos a ocorrerem no Verão e os mais elevados no Inverno –, este padrão modificou-se substancialmente nos últimos dois anos.
Com o surgimento da covid-19, mesmo no Verão o decréscimo de doentes foi brutal. E no Inverno, as quedas foram completamente atípicas. Aliás, os dois piores meses da pandemia – Janeiro e Fevereiro de 2021, com 10.137 e 10.457 internados, respectivamente – coincidiram com os mais baixos números de internados por doenças respiratórias: para aqueles dois meses foram de apenas 4.396 e 3.558, respectivamente.
Se se comparar o número de internados por doenças respiratórias nos dois primeiros meses de 2021 – um total de 7.954 – com os internados nos meses de Janeiro e Fevereiro 2017 – com um surto gripal relevante, que levou à hospitalizações de 25.821 pessoas –, fica-se com uma ideia clara do impacte ao nível da pressão hospitalar do “desaparecimento” da gripe durante a pandemia.
No entanto, a pandemia aliviou fortemente outras áreas hospitalares como foram sobretudo os casos das unidades de tratamento de doenças do aparelho circulatório e digestivo e também de neoplasias (cancros).
Segundo os dados do SNS, confrontando o período 2018-2019 com 2020-2021, houve menos 37.800 internados (redução de 15,7%) por doenças do aparelho circulatório, menos 34.443 internados (redução de 19,5%) por doenças do aparelho digestivo e menos 30.759 internados (redução de 17,4%) por neoplasias.
Número total de internados por grupo de doenças nos biénios 2018-2019 e 2020-2021. Cálculos e análise: PÁGINA UM.
O cenário, contudo, foi generalizado para todas as doenças e afecções, mesmo até em internamentos por lesões, acidentes, transtornos mentais e doenças dos olhos. Na análise realizada pelo PÁGINA UM, observam-se nove grupos de doenças com reduções superiores a 20%
Esse efeito observou-se mesmo nos internamentos relacionados com a gravidez (menos 12,4%), malformações congénitas e similares (menos 17,8%) e condições originadas no período perinatal (-29,0%), mas aí a causa foi outra: as opções da estratégia política do Governo que resultou numa incerteza económica que retraiu os casais na decisão de terem filhos.
São 14 os gráficos. O PÁGINA UM faz nova análise à base de dados da morbilidade e mortalidade do Portal da Transparência do Serviço Nacional de Saúde (SNS), desta vez pesquisando um indicador fundamental da pressão hospitalar: o número de dias de internamentos por mês desde 2017. Vimos cada um dos hospitais… em, pois bem, durante os dois primeiros anos da pandemia, houve muitos hospitais do SNS, a começar pelos de Lisboa, que nunca tiveram tanto “descanso”. O Ministério da Saúde fez-nos crer o contrário.
A pressão hospitalar sempre foi tema quente desde a chegada da pandemia da covid-19 a Portugal. Está de novo na hora do dia, graças a um histriónico director das urgências do Hospital de São João do Porto a pré-anunciar uma catástrofe apenas por um ligeiro acréscimo no fluxo de doentes nas últimas semanas. Como noutras ocasiões, desde Março de 2020.
A mensagem política e social ao longo da pandemia foi sempre no sentido de a covid-19 não sobrecarregar os serviços hospitalares e sobretudo as urgências, num país com mais de um milhão de pessoas sem médico de família.
Ficaram mesmo célebres as ambulâncias em frente ao Hospital de Santa Maria. Meta-se as palavras “hospitais” e “entupidos” no Google News, e encontraremos uma lista de notícias, umas mais clássicas anteriores à pandemia, outras mais recentes, de 2020, 2021 e mesmo deste ano.
Mas será isto mesmo verdade? Teremos médicos, enfermeiros e mesmo administradores hospitalares à beira da exaustão, como se nada houvesse parecido àquilo que sucedeu nos últimos dois anos.
Fomos fazer contas à vida nos locais onde se evita a morte. Melhor dizendo, o PÁGINA UM foi analisar, em detalhe, um dos indicadores fundamentais da pressão hospitalar: o número de dias acumulados de internamento nas unidades do Serviço Nacional de Saúde (SNS).
Os dias de internamento constituem não um retrato, como sucede nos picos conjunturais de afluência, mas sim um balanço de um período alargado (geralmente, um mês, um trimestre, um ano), permitindo aferir se existe uma variação relevante na procura de recursos materiais e humanos (médicos, enfermeiros, técnicos de saúde e auxiliares) para tratamento de doentes em situação mais delicada.
Para isso, recorremos a dados indesmentíveis – ou, pelo menos não desmentíveis, porque oficiais – pelo Ministério da Saúde ou pela imprensa que segue a “espuma dos dias”: a base de dados da morbilidade e mortalidade hospitalar do Portal da Transparência do Serviço Nacional de Saúde (SNS).
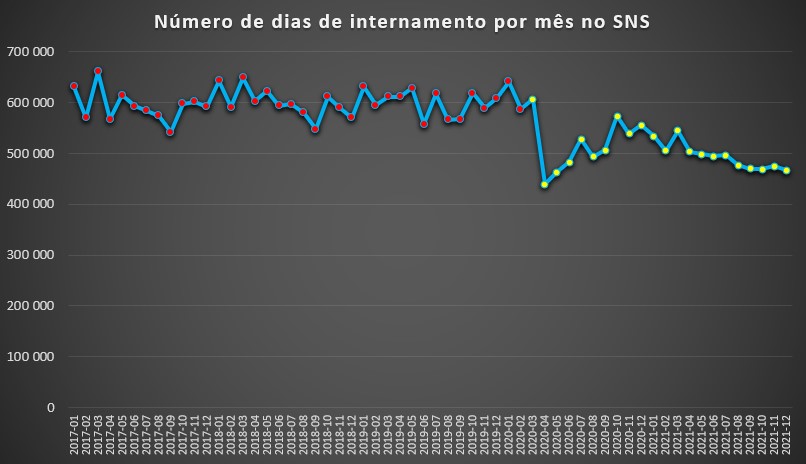
Independentemente de ter ocorrido, efectivamente, uma maior pressão em diversos sectores hospitalares – sobretudo dos profissionais de saúde que estiveram alocados ao tratamento dos doentes-covid, em grande parte pela maior logística e disponibilidade que exigiu –, os dados do SNS afinal revelam, globalmente, uma evidência que destoa da visão mais “popular”: desde 2017 – período a partir do qual existem registos mensais contabilizando dias acumulados de internamento –, o ano com menor pressão foi o ano passado.
Com efeito, no total dos 12 meses de 2021, contabilizaram-se 5.931.618 dias de internamento, o que contrasta com 6.411.908 dias registados em 2020, que incorporou 10 meses (Março a Dezembro) já afectados pela pandemia. Se considerarmos a média mensal de 2020-2021 (cerca de 514.313 dias de internamento) com a de 2017-2019 (597.694 dias), a queda é de 14%.
Na verdade, antes da pandemia, a pressão hospitalar – medida pela “procura” (ou ocupação) de camas para internamento –, mantinha-se mais ou menos estável, até porque dependia da disponibilidade dos hospitais, tanto em camas “físicas” como em pessoal para tratar os doentes.
De acordo com a base de dados do SNS, os dias de internamento por mês entre Janeiro de 2017 até Fevereiro de 2020 – em vésperas da chegada do SARS-CoV-2 a Portugal – variavam entre os 540 mil e os 660 mil, com os valores mais elevados a ocorrerem, geralmente, no primeiro trimestre de cada ano.
Porém, com a chegada da pandemia em Março de 2020, tudo mudou, sobretudo no mês seguinte, onde quem estava doente via um hospital como se fosse o diabo a ver uma cruz: fugia. Por esse motivo, Abril de 2020 nem sequer contabilizou 440 mil dias de internamento, uma descida de quase 27% face ao mês anterior.
Número de dias de internamento em cada mês na totalidade das unidades do Serviço Nacional de Saúde desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Nos meses seguintes registar-se-ia um ligeiro aumento do número de dias de internamento, até que em Outubro de 2020 a pressão hospitalar atingiu valores considerados normais.
Contudo, com a intensificação da pandemia, e sobretudo com o surgimento da pior fase da covid-19 entre Dezembro de 2020 e Fevereiro de 2021, registou-se nova redução neste indicador.
Mas passada a tempestade, e apesar da manutenção das restrições, justificadas em parte para não sobrecarregar os hospitais com doentes-covid, o número de dias de internamento foram suavemente diminuindo. E mesmo com a chegada do último Inverno. Na verdade, desde Maio de 2021 o número acumulado de dias de internamento estiveram sempre abaixo dos 500 mil por mês. Ora, entre 2017 e 2019 contabilizam-se 16 meses acima de 600 mil dias de internamento.
As realidades foram, contudo, bastante distintas de unidade de saúde para unidade de saúde.
Para fazer uma análise mais fina, o PÁGINA UM seleccionou os centros hospitalares ou hospitais que, entre 2017 e 2019, registaram mais de 500 mil dias de internamento. De forma clara, evidenciam, em quase todos, um impacte brutal entre Março e Abril de 2020, no período de maior pânico, mas as evoluções são depois muito distintas.
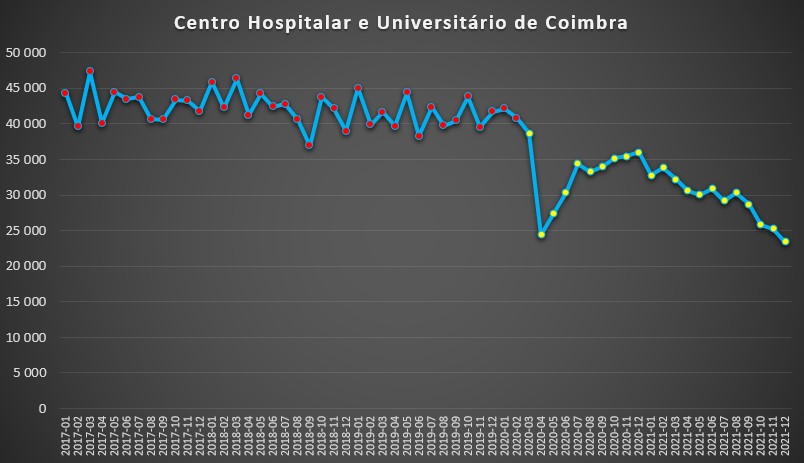
Por exemplo, no Centro Hospitalar e Universitário de Coimbra, a queda no internamento foi impressionante. Antes da pandemia, raramente havia um mês com menos de 40 mil dias de internamento. Em Abril de 2020 desceu para apenas 24.393 dias, uma redução da ordem dos 40%.
Número de dias de internamento em cada mês no Centro Hospitalar e Universitário de Coimbra desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Ao longo de 2020, a pressão hospitalar, medida por este indicador, aumentou mas nunca chegou ao patamar da “normalidade” anterior à pandemia. Em Dezembro ultrapassou-se ligeiramente os 36 mil dias. Em seguida, ao longo de todo o ano de 2021, observou-se uma tendência de decréscimo, bastante acentuada mês após mês, de sorte que, em Dezembro do ano passado, se atingiu um novo mínimo desde 2017: somente 23.468 dias de internamento.
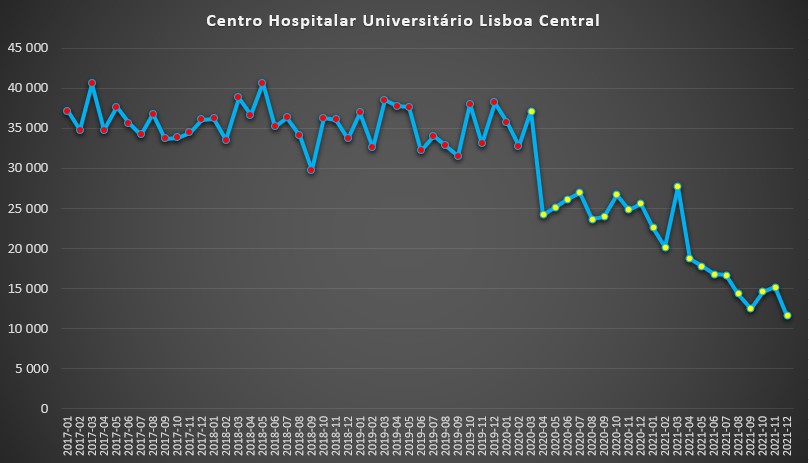
Situação ainda mais drástica observou no Centro Hospitalar de Lisboa Central – que agrega, entre outros, os hospitais de São José, D. Estefânia, Curry Cabral e Santa Marta. Com número de dias de internamento por mês a situar-se, geralmente, entre os 30 mil e os 40 mil antes da pandemia, a quebra foi bastante acentuada logo em Abril de 2020: contabilizaram-se um pouco menos de 25 mil. Uma queda de quase 33% face ao mês anterior.
Número de dias de internamento em cada mês no Centro Hospitalar Universitário de Lisboa Central desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Até Dezembro desse ano, o indicador manteve-se sempre em redor dos 25 mil dias, iniciando depois nova queda acentuada, com excepção de Março de 2021. O mês de Dezembro do ano passado foi um período nunca visto desde 2017: somente 11.566 dias de internamento. Face ao máximo mensal registado no período (40.609 dias em Março de 2018), significa uma queda de 72%.
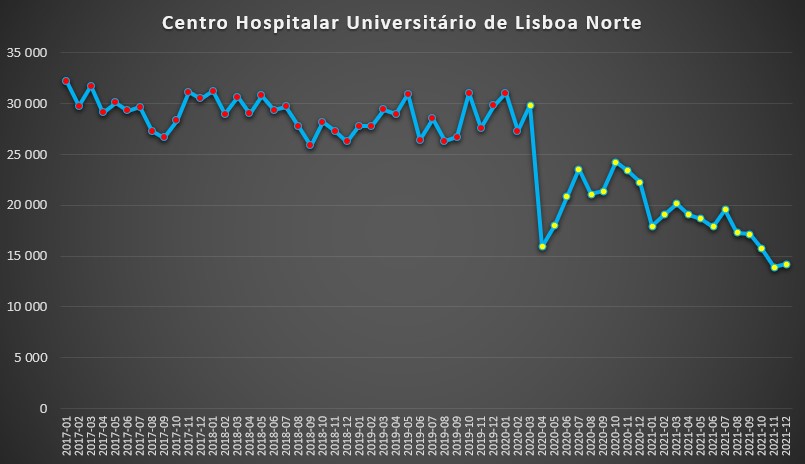
O padrão do “vizinho” Centro Hospitalar Universitário de Lisboa Norte – que integra, entre outros, os hospitais de Santa Maria e Pulido Valente – foi idêntico, embora com uma queda ainda mais acentuada após a chegada do SARS-CoV-2, que trouxe uma debandada geral às unidades de saúde.
Número de dias de internamento em cada mês no Centro Hospitalar Universitário de Lisboa Norte desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Se antes da pandemia, o fluxo de internamentos mensais situava-se entre os 25 mil e os 32 mil, em Abril de 2020 decaiu para baixo dos 16 mil, um tombo de 47% face ao mês anterior. Houve depois uma “recuperação” nos meses seguintes até Outubro desse ano, mas seguiu-se uma acentuada tendência de descida nos dias de internamento, incluindo mesmo nos meses de pico da pandemia do Inverno de 2020-2021, quando o Hospital de Santa Maria era palco mediático de ambulâncias em fila indiana para “despejar” doentes. Problemas de logística, na verdade. Os últimos dois meses de 2021 ficaram ambos abaixo dos 15 mil dias de internamento – valores que representam cerca de metade da “normalidade” pré-pandemia.
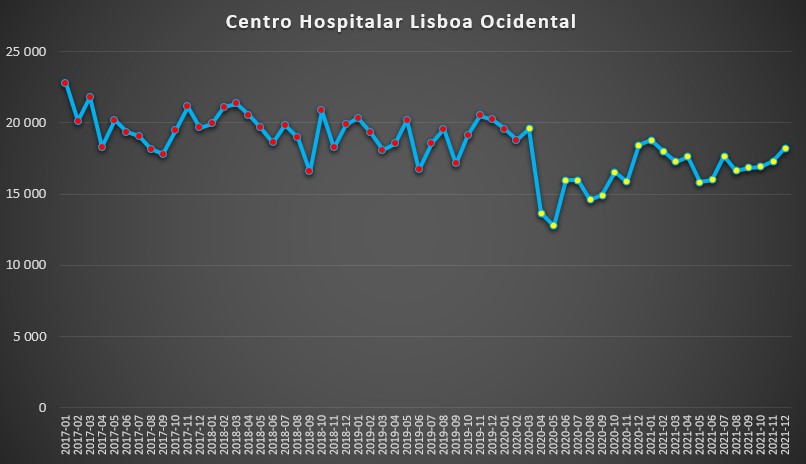
O Centro Hospitalar Lisboa Ocidental destoa deste padrão. Houve efectivamente uma queda abrupta entre Março e Abril de 2020, mas rapidamente se passou para um padrão de “normalidade” pré-pandemia, sobretudo ao longo do ano passado. Porém, até Dezembro de 2021 nunca se chegou a ultrapassar os 20 mil dias de internamento em qualquer mês, algo que sucedeu por vezes no período 2017-2019.
Número de dias de internamento em cada mês no Centro Hospitalar Universitário de Lisboa Ocidental desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
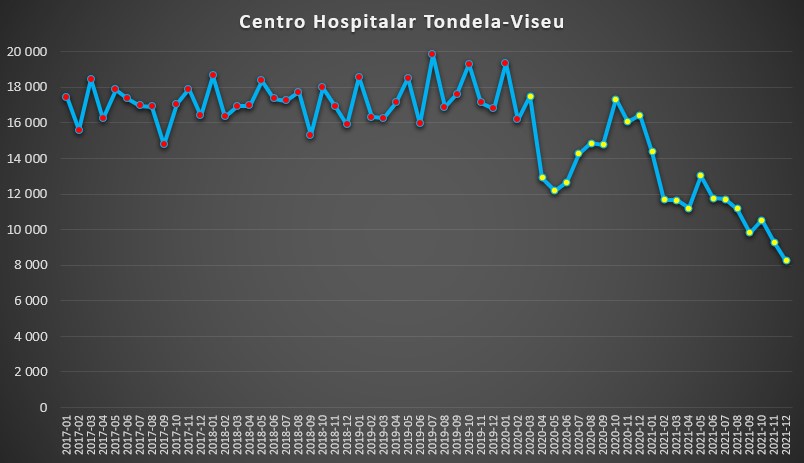
Fora dos grandes centros urbanos, alguns centros hospitalares também mostraram este padrão. Foi o caso do de Tondela-Viseu. Neste caso, a queda de dias de internamento no início da pandemia foi mais curto – apenas entre Abril e Setembro de 2020 –, mas quando se pensava que os valores deste indicador começariam a estar próximos da “normalidade”, houve nova e mais persistente queda. No último mês do ano passado contabilizaram-se apenas 8.241 dias de internamento, uma queda de 58% face ao mês com maior pressão hospitalar desde 2017 (Julho de 2019).
Número de dias de internamento em cada mês no Centro Hospitalar Tondela-Viseu desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
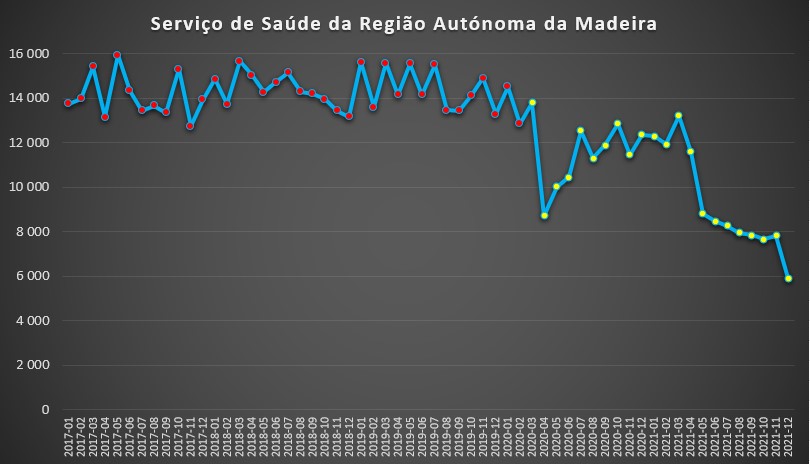
Muito parecida foi a evolução das unidades de saúde na Região Autónoma da Madeira: uma descida abrupta em Abril de 2020, seguindo-se uma “recuperação” para níveis próximos da “normalidade” pré-pandémica, que perdurou até Março de 2021. A partir desse mês registou-se uma queda acentuadíssima, com o valor de Dezembro do ano passado a rondar apenas os seis mil dias de internamento.
Número de dias de internamento em cada mês nas unidades de saúde da Madeira desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
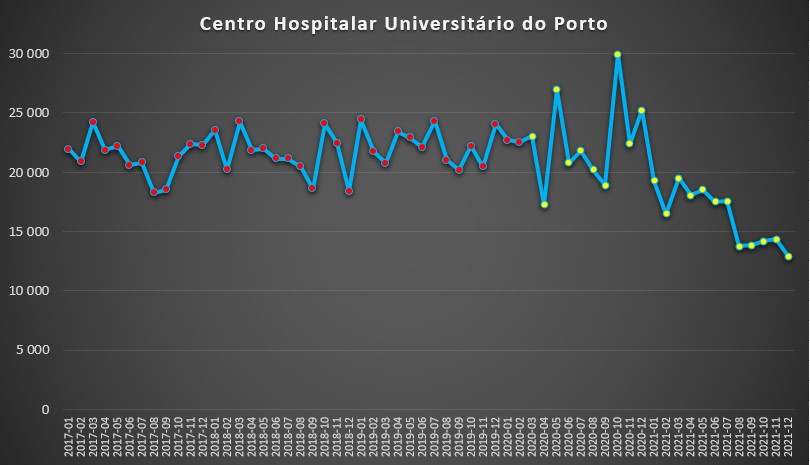
No concelho do Porto observaram-se duas situação muito díspares. No Centro Hospitalar Universitário do Porto, que integra o Hospital de Santo António, registou-se mesmo, após o “susto” inicial de Abril de 2020, um aumento da média de dias de internamento, com um pico em Outubro daquele ano. Porém, a partir de Agosto do ano passado, este indicador desceu para valores bastante baixos, inferiores a 15 mil dias de internamento.
Número de dias de internamento em cada mês no Centro Hospitalar Universitário do Porto desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
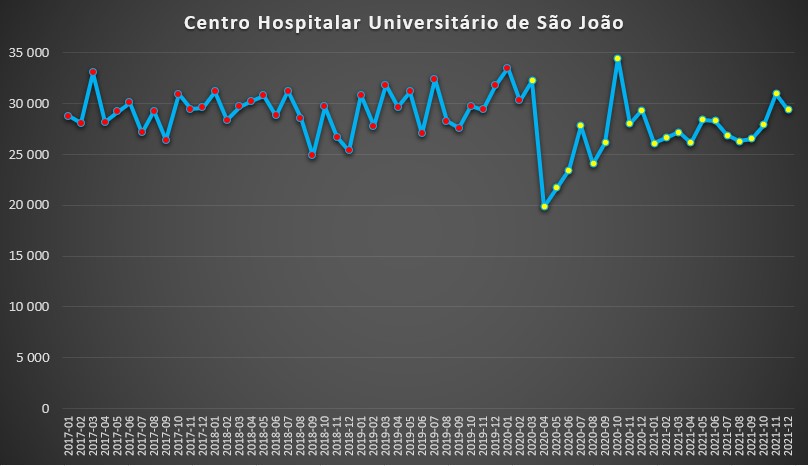
Já no outro hospital da cidade do Porto, o do São João, o cenário foi bastante diferente: apenas os meses de Abril, Maio e Junho de 2020 se registaram dias de internamento abaixo da “normalidade” pré-pandemia, ficando depois sempre em valores sensivelmente idênticos ao período 2017-2019.
Número de dias de internamento em cada mês no Centro Hospitalar Universitário de São João desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
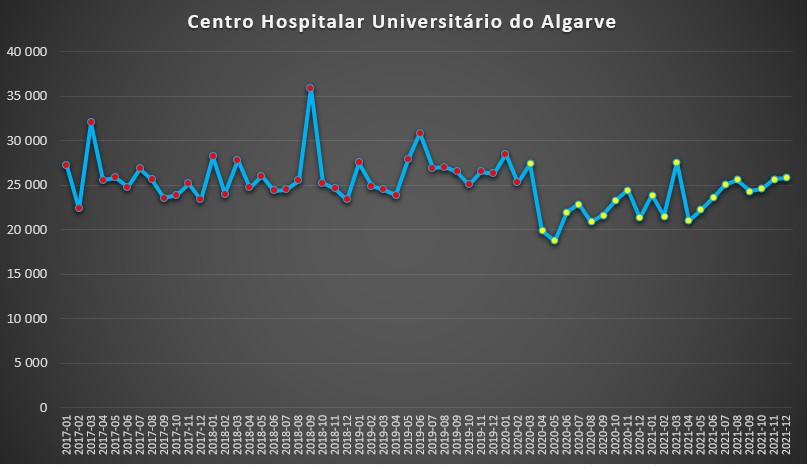
Na mesma linha se encontra o Centro Hospitalar Universitário do Algarve – que integra os hospitais de Faro, Portimão e Lagos –, que após uma repentina descida de um pouco mais de 20% nos dias de internamento entre Março e Abril de 2020, foi depois caminho para uma “normalidade” pré-pandémica.
Número de dias de internamento em cada mês no Centro Hospitalar Universitário do Algarve desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
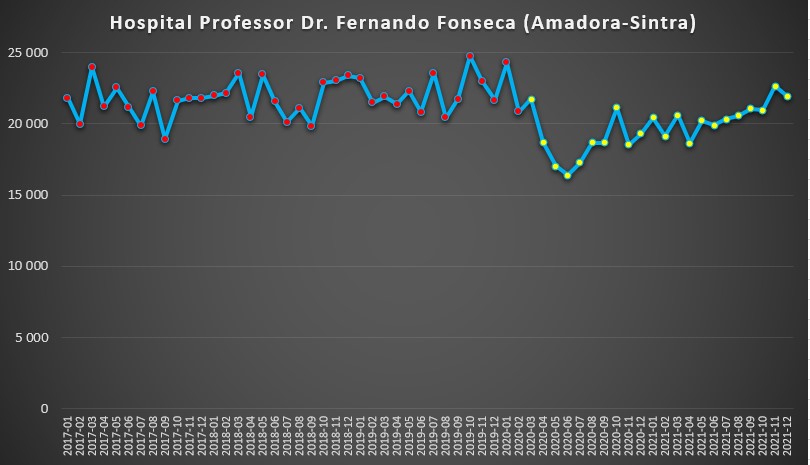
No Hospital Amadora-Sintra a situação também foi quase similar. Se antes da pandemia o número de dias de internamento se situava quase sempre entre os 20 mil e 25 mil dias por mês, a emergência da covid-19 provocou um abaixamento nas hospitalizações, com o mínimo a ser atingido em Junho de 2020 (16.381 dias). A partir desse mês, o crescimento tem sido gradual, mas ainda não chegou sequer aos 23 mil dias de internamento.
Número de dias de internamento em cada mês no Hospital Amadora-Sintra desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
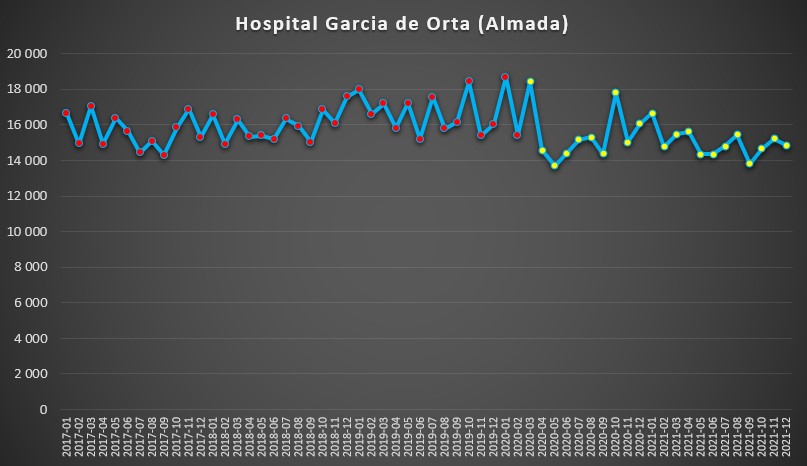
No Hospital Garcia de Orta, em Almada, a pandemia não trouxe alterações relevantes na pressão hospitalar. É certo que houve uma queda percentualmente relevante logo no início da pandemia (entre Março e Abril de 2020), mas a a seguir as variações estão dentro de um padrão de “normalidade”.
Número de dias de internamento em cada mês no Hospital Garcia de Orta (Almada) desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
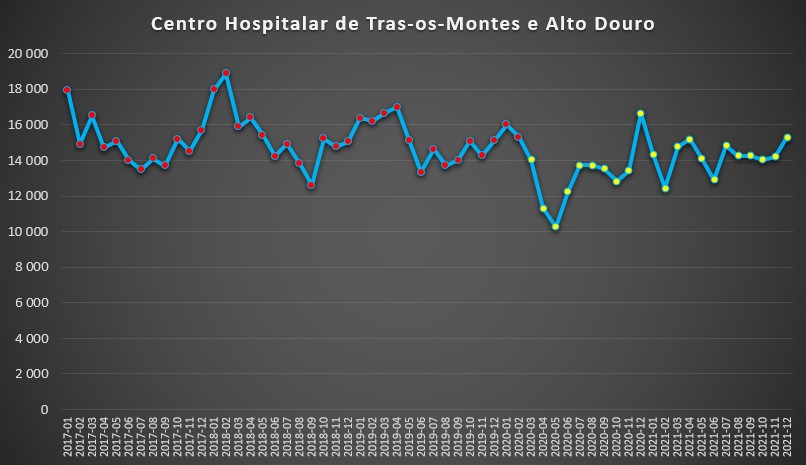
Situação semelhante se viveu no Centro Hospitalar de Trás-os-Montes e Alto Douro – que integra os hospitais de Vila Real, Chaves e Lamego. Houve, efectivamente, uma queda na pressão hospitalar acentuada nos primeiros meses da pandemia, passando de cerca de 14 mil dias de internamento em Março de 2020 para 10 mil em Maio daquele ano, mas depois os valores regressaram aos padrões de “normalidade”.
Número de dias de internamento em cada mês no Centro Hospitalar de Trás-os-Montes e Alto Douro desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
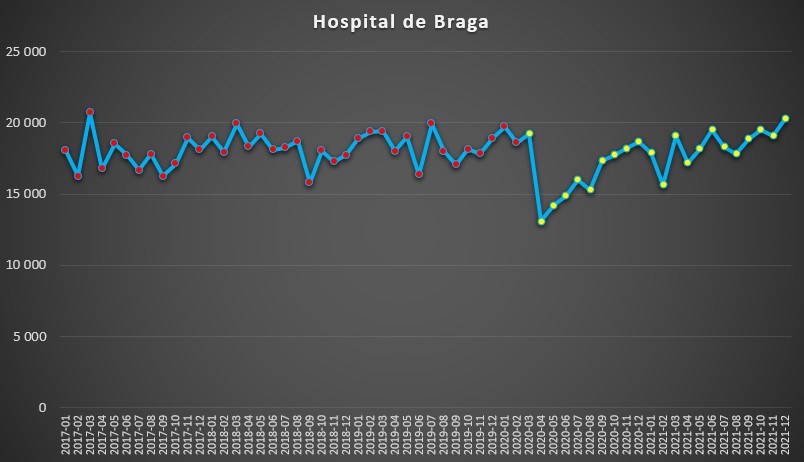
Por fim, o Hospital de Braga foi o único, de entre os seleccionados, que acabou o ano de 2021 com níveis de pressão, medidos em termos de dias de internamento, ligeiramente acima do “normal” antes da pandemia. No mês de Dezembro do ano passado registaram-se 20.294 dias de internamento, o segundo valor mensal mais elevado desde 2017.
No entanto, o percurso deste hospital nortenho foi semelhante aos demais com a chegada da pandemia: queda abrupta dos internamentos entre Março e Abril de 2020. Depois continuou sempre em crescimento até atingir valores ligeiramente acima da “normalidade”.
Número de dias de internamento em cada mês no Hospital de Braga desde 2017 até 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Em suma, a análise do PÁGINA UM mostra, com base em dados oficiais, que foi criado um mito em redor de uma alegada existência de uma pressão hospitalar incomportável criada pela pandemia, e que justificaria restrições sociais e a suspensão de operações programadas.
Na verdade, uma das explicações para esta quebra nos internamentos em grande parte dos hospitais será mesmo a redução de intervenções cirúrgicas programadas, e que resultariam em internamentos de recuperação. Quais as consequências destes adiamentos? O Ministério da Saúde poderá, certamente, responder. Ou melhor, deveria responder.
Os especialistas em oncologia têm estado a alertar para a elevada probabilidade de um aumento significativo de mortes por cancros devido à instabilidade e decisões do Serviço Nacional de Saúde (SNS) durante a pandemia, que levou à redução dos rastreios, diagnósticos e tratamentos. Porém, a análise do PÁGINA UM à base de dados da morbilidade e mortalidade do Portal da Transparência do SNS mostra um surpreendente paradoxo: nunca como nos últimos meses se morreu tão pouco nos hospitais por causa de cancros. Ou os doentes terminais andam a ser enviados para casa ou há embuste…
Em cerca de dois anos de presença da covid-19 em Portugal, não cessaram os alarmes nos últimos meses sobre as consequências da gestão da pandemia nos atrasos nos diagnósticos de cancros. No final do ano passado, a Organização Europeia contra o Cancro estimou que mais de 100 milhões de rastreios não se tinham realizado ao longo de 2020 e 2021 no Velho Continente.
Em Portugal, os especialistas na área Oncologia têm alertado para a iminência de uma “pandemia” de cancros, e de mortes, por via da suspensão e atraso de rastreios e diagnósticos, tanto por razões políticas como pelo medo de muitas pessoas em frequentarem unidades de saúde.
Porém, Portugal é um país suigeneris. De acordo com a base de dados da morbilidade e mortalidade hospitalar, disponível no Portal da Transparência, o mês com menos mortes causadas por neoplasias foi Janeiro deste ano, o último com informação desde 2017. Mas este não foi caso esporádico.
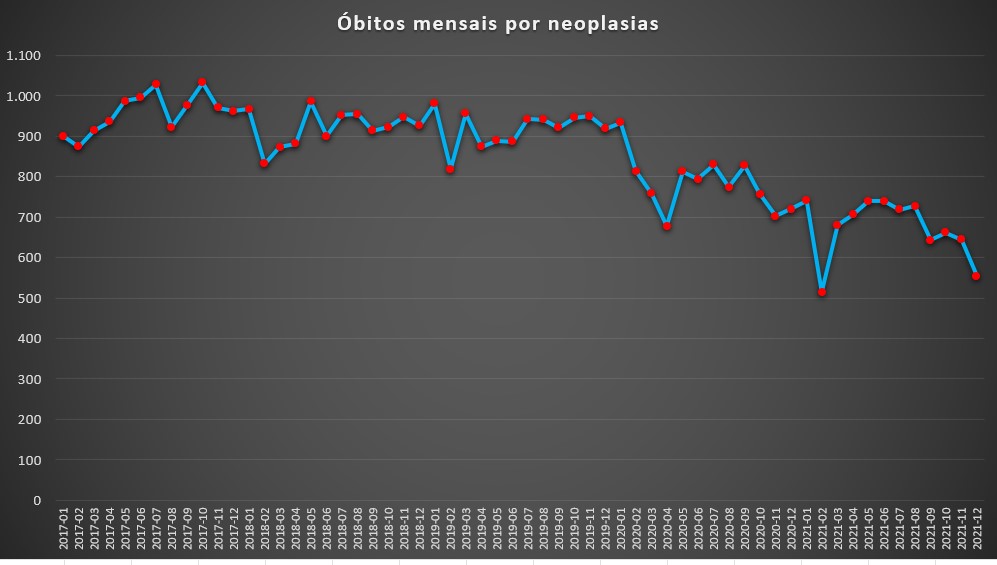
De acordo com a análise da informação realizada pelo PÁGINA UM, a redução da mortalidade causada pelos mais diversos cancros tem sido anormalmente baixa desde o início da pandemia da covid-19, em Março de 2020. Com efeito, no período pré-pandemia – e desde Janeiro de 2017, data do início do registo –, os óbitos em meio hospitalar por neoplasias situavam-se entre os 800 e os 1050 por mês. Ou seja, sem grandes oscilações.
Geralmente, os valores ligeiramente mais baixos observavam-se no Inverno, mas por uma razão simples: devido à fragilidade de muitos doentes oncológicos, muitas mortes são “antecipadas” por outro tipo de doenças, sobretudos infecções respiratórias como as pneumonias. Ora, tal significava que as doenças respiratórias acabavam por ser consideradas, em alguns casos, a causa do óbito, e não os cancros.
Em todo o caso, com a chegada da covid-19 em território português, as mortes por cancro tiveram uma queda acentuada. Em Março de 2020, os óbitos desceram para 758. Comparando com os meses homólogos do período anterior à pandemia foi uma descida significativa: em 2017 tinham morrido 914, em 2018 foram 873 e em 2019 situaram-se nos 955.
Óbitos totais por mês, por neoplasias, registados nas unidades do SNS entre Janeiro de 2017 e Dezembro de 2021. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Em Abril de 2020, os óbitos por cancros registados em meio hospitalar ainda desceram mais: somente 678. Nos meses seguintes, apesar de os valores subirem ligeiramente nunca superaram os 830 óbitos.
Com o Outono e Inverno de 2020-2021 – que marcaria o período mais crítico da pandemia, com as mortes por covid-19 a subirem, atingindo, em alguns dias de Janeiro valores a rondarem os 300 óbitos –, os desfechos fatais atribuídos aos cancros reduziram ainda mais. No período compreendido entre Novembro de 2020 e Janeiro de 2021, óbitos mensais situaram-se entre os 700 e os 750. No total, neste trimestre registaram-se 2.173 óbitos por cancro, uma descida de 22% em relação ao período homólogo anterior.
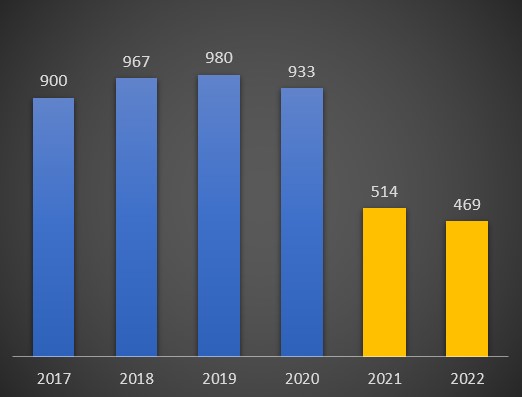
No mês de Fevereiro do ano passado, a queda ainda foi mais notória: 514 óbitos, um valor perfeitamente atípico. Nos meses seguintes, o padrão de anormalmente baixa mortalidade por cancros manteve-se. Sempre abaixo dos 750 óbitos até Agosto, e a partir de Setembro ainda mais baixo. No último mês do ano passado, em Dezembro, as mortes por cancro nas unidades de saúde foi de 554. E em Janeiro deste ano situar-se-ia nos 469 óbitos. Note-se que, nos anos anteriores à pandemia, esta doença matou 900, 967, 980 e 933 pessoas, respectivamente no primeiro mês dos anos de 2017, 2018, 2019 e 2020 – ou seja, antes da pandemia.
Óbitos por neoplasias registados no mês de Janeiro entre 2017 e 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
O absurdo está assim instalado em Portugal, e será provável que se mantenha, excepto se o Ministério da Saúde esclarecer este paradoxo, que se pode caricaturar: a pandemia “eliminou” mortes por cancro.
Obviamente, na verdade, haverá duas possíveis explicações, que poderão estar conjugadas, mas em qualquer dos casos são graves.
Por um lado, um número muito significativo de doentes oncológicos terminais tiveram – e, provavelmente, em muitos casos de forma injustificada – a covid-19 como causa de morte, inflacionado o impacte da pandemia. E, dessa forma, também de forma injustificada, a estatística dos cancros está enviesada, por subestimada.
Note-se que, nos três anos anteriores à pandemia, as neoplasias causavam por ano cerca de 11 mil óbitos, sem grandes variações, o que é normal face aos padrões epidemiológicos das doenças oncológicas em Portugal. Porém, em 2020 (com nove meses em pandemia) desceu para os 9.398 óbitos, e decaiu ainda mais em 2021: apenas 8.067 – uma descida de 28% face ao triénio anterior à pandemia. São mais de três mil mortes a menos.
No entanto, como estas estatísticas se referem somente aos óbitos registados em meio hospitalar – e, portanto, não se inclui as mortes de doentes oncológicos ocorridas em residências e lares –, poder-se-á sempre dizer – à falta da divulgação de dados oficiais pela Direcção-Geral da Saúde (DGS), apesar da existência do Sistema de Informação dos Certificados de Óbitos (SICO) – que os cancros passaram a dizimar menos nos hospitais, porque os doentes terminais foram enviados para casa.
Mas, se assim fosse – e significando assim que se abandonariam muito doentes à sua sorte nos derradeiros momentos de vida, o que parece pouco provável do ponto de vista humano –, deveriam então esses dados ser fornecidos de forma clara e transparente, permitindo avaliações independentes sobre o verdadeiro impacte da pandemia na evolução dos cancros.
Se assim não for, se não houver transparência, se o obscurantismo continuar a imperar, uma coisa é certa: o Governo vai anunciar daqui a uns tempos, com pompa e circunstância, que nunca como antes os problemas oncológicos estiveram controlados.
E que o Governo conseguiu recuperar todos os atrasos no rastreio, nos diagnósticos e no tratamento dos cancros. E a Estatística, se o Governo quiser, dirá que as pessoas, de facto, até morrem menos de cancro. Morreram de outras coisas, e cada vez mais, mas não de cancro… E isso será o embuste em todo o seu esplendor.
O PÁGINA UM começa, a partir de hoje, a apresentar um conjunto de análises à base de dados da morbilidade e mortalidade do Portal da Transparência do Serviço Nacional de Saúde (SNS). Neste primeiro artigo revela-se que, afinal, houve muitas mortes por covid-19 que “escaparam” a tratamento hospitalar, e que a pressão sobre o SNS foi, na verdade, com excepção de um curto período (Dezembro de 2020-Fevereiro de 2021), atipicamente baixo nos dois anos de pandemia. E essa situação mostra-se evidente sobretudo a partir de Março do ano passado.
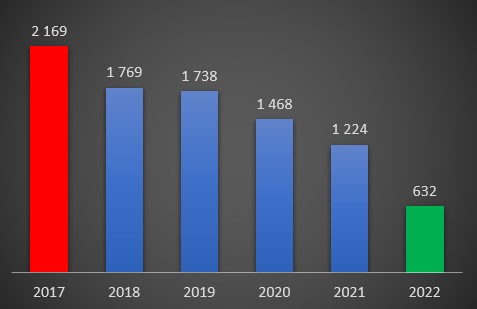
São dados oficiais. Indesmentíveis pelo Ministério da Saúde. Os registos da morbilidade e mortalidade hospitalar do Portal da Transparência do Serviço Nacional de Saúde (SNS) revelam que, durante os dois anos da pandemia (2020-2021) morreram afinal menos pessoas nos hospitais portugueses do que nos dois anos anteriores (2018-2019).
E apesar de a covid-19 ter constituído um factor de mortalidade importante (12% dos óbitos nas unidades hospitalares) em 2020 e 2021, estranhamente, ou talvez não, uma parte relevante de doenças mortais acabaram por registar fortes quedas.
A análise do PÁGINA UM a esta base de dados do SNS – com informação detalhada por mês e mesmo por unidade de saúde, incluindo internamentos e óbitos ocorridos em unidades de saúde – desencadeia uma reflexão sobre a forma como decorreu a estratégia política de gestão da pandemia.
Nessa medida, vale a pena olhar para a evolução do registo mensal das mortes em meio hospitalar – que, sem prejuízo do aspecto humano relevante, ademais sabendo-se que houve um acréscimo importante de óbitos fora das unidades de saúde –, pois constitui sempre um indicador fundamental em termos de Saúde Pública. Neste caso, nem que seja por permitir aferir indirectamente o grau de pressão e complexidade dos casos a que sujeita o SNS e os seus profissionais.
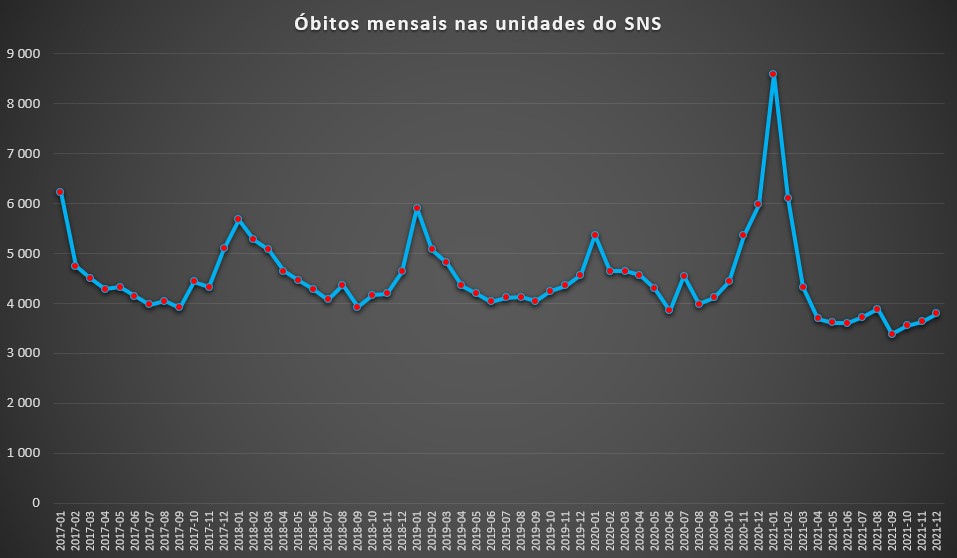
Ora, aquilo que se verificou – pegando nos registos das mortes por todas as causas ocorridas em meio hospitalar – é que, com excepção de um curto período, entre Dezembro de 2020 e Fevereiro de 2021, o SNS não denotou uma sobrecarga. No caso de Janeiro de 2021 houve mesmo um evidente colapso com um recorde de 8.590 óbitos. No período anterior à pandemia – e desde 2017, com informação na base de dados do SNS –, nos piores meses contabilizavam-se cerca de seis mil óbitos em meio hospitalar, sobretudo no mês de Janeiro, estando associado aos surtos gripais (causadores de mais mortes por doenças respiratórias) e ao frio (adjuvante de doenças mortais do aparelho circulatório).
Óbitos totais por mês, por todas as causas, registados nas unidades do SNS entre Janeiro de 2017 e Dezembro de 2021. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Porém, excluído esse trimestre, ninguém que agora surgisse por aqui sem saber que houve uma pandemia poderia dizer que o SNS esteve sob pressão. Pelo contrário: desde Março do ano passado, o número de óbitos mensais registados nos hospitais do SNS foi sempre atipicamente baixo (sempre abaixo dos 4.000). E esta situação observou-se sobretudo com chegada das estações associadas a uma maior mortalidade (Outono e Inverno). Por exemplo, em Dezembro passado apenas se contabilizaram 3.793 óbitos. No mês homólogo dos três anos anteriores à pandemia, os óbitos em meio hospitalar foram muito superiores: 5.089 em 2017, 4.637 em 2018 e 4.561 em 2019.
O mês de Janeiro deste ano – que já consta da base de dados do SNS – surge com 3.461 óbitos, um valor extraordinariamente baixo, tanto mais que chega a ser inferior aos meses de Verão pré-pandemia.
Mas uma análise mais detalhada desta base de dados suscita ainda mais perplexidades, e muitos questionamentos.
E a começar pelo número de mortes causadas pela própria covid-19. Apesar de ter sido considerada uma doença de elevada infecciosidade – que obrigou, na esmagadora maioria dos casos ao internamento de casos moderados e graves –, constata-se que, afinal, morreram nas unidades de saúde até Dezembro de 2021 um total de 12.837 pessoas devido à acção directa do SARS-CoV-2. Este valor é “apenas” 68% do total dos óbitos contabilizados pela Direcção-Geral da Saúde (DGS) em 2020 e 2021. Ou seja, dos 18.974 óbitos por covid-19 contabilizados até 31 de Dezembro de 2021, houve 6.137 que faleceram fora de unidades de saúde, em lares ou nas suas residências.
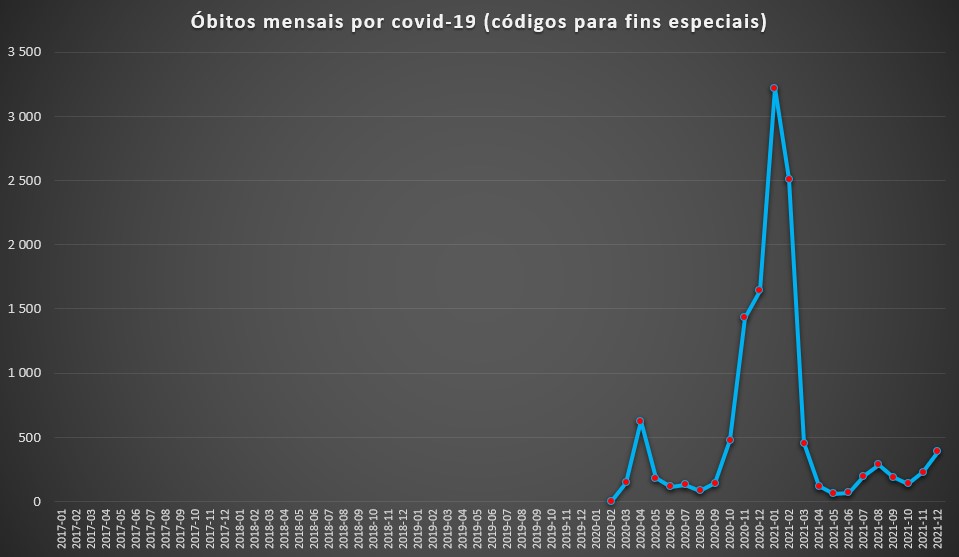
Óbitos totais por mês, causados por covid-19 (integrados no grupo “Códigos para fins especiais), registados nas unidades do SNS entre Janeiro de 2017 e Dezembro de 2021. Fonte: SNS. Cálculos e análise: PÁGINA UM.
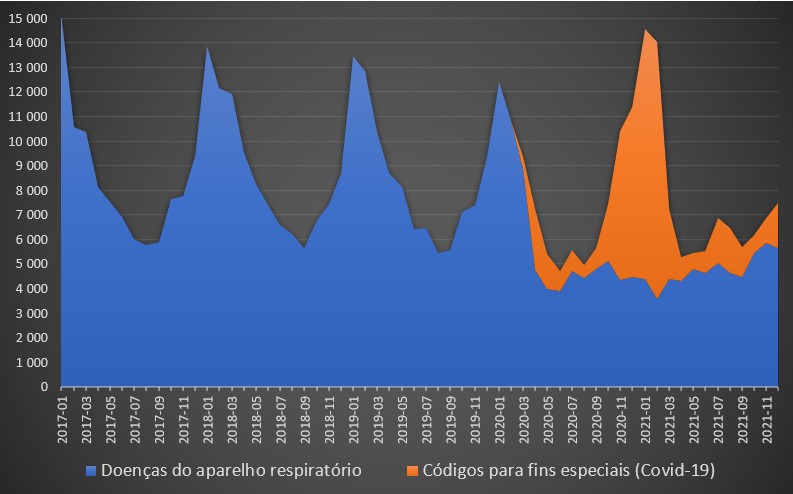
Saliente-se que, na base de dados do SNS, a covid-19 não surge explicitamente como a causa de morte, mas no grupo das doenças catalogada em “Códigos para fins especiais”. A covid-19 e sequelas associadas (código U) são praticamente as únicas doenças mortais inseridas neste grupo, razão pela qual apenas começaram a surgir a partir de Março de 2020.
Nesse mês, oficialmente morreram nos hospitais portugueses 147 pessoas com esta doença, chegando às 626 no mês seguinte. O período mais negro surgiu, como conhecido, entre Novembro de 2020 e Fevereiro de 2021: no primeiro mês deste período morreram 1.431, em Dezembro 1.643, em Janeiro 3.320 e em Fevereiro 2.512.
Até final de 2021, em mais nenhum mês se ultrapassaram os 500 óbitos. Em Janeiro deste ano – quando se registou uma vaga de casos positivos, com quase 1,3 milhões de casos –, nos hospitais morreram 560 pessoas por covid-19. No entanto, a DGS anunciou, para esse mês, um total de 1.002 óbitos, o que significa que 44% terão falecido fora de unidades de saúde. Ou então os números terão sido empolados.
Se causa estranheza esta relevante discrepância entre óbitos por covid-19 em meio hospitalar e fora das unidades de saúde – o que significará que muitos casos graves causados pelo SARS-CoV, susceptíveis de serem (como foram) letais, não terão assim tido tratamento hospitalar –, maior estupefacção surge quando se confronta a mortalidade por grupos de doenças durante a pandemia com o período homólogo anterior.
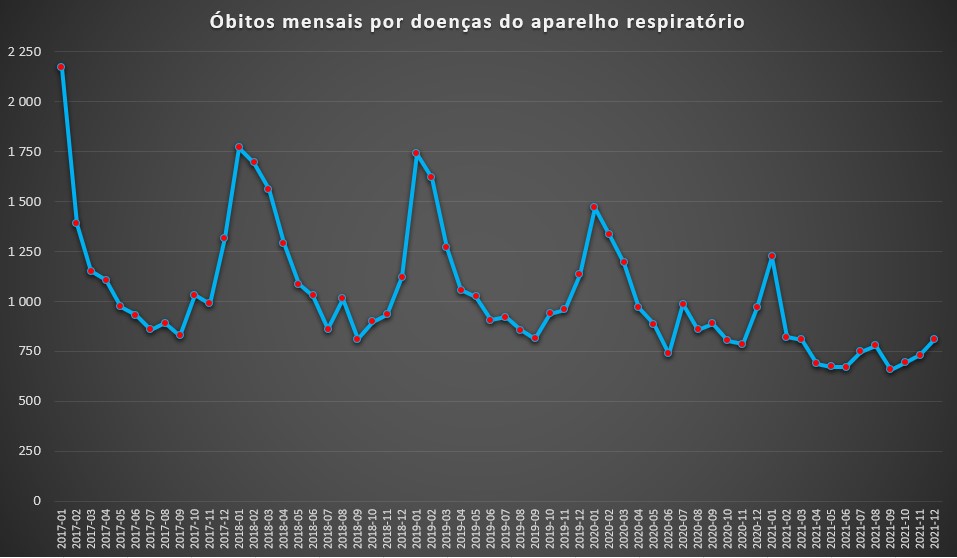
Nesta primeira parte, analisamos primeiro as doenças respiratórias – que, supostamente, “beneficiaram” do desaparecimento da gripe e, segundo a DGS, das medidas não-farmacológicas.
Óbitos totais por mês, causados por doenças do aparelho respiratório, registados nas unidades do SNS entre Janeiro de 2017 e Dezembro de 2021. Fonte: SNS. Cálculos e análise: PÁGINA UM.
No período 2018-2019, segundo a base de dados do SNS, contabiliza-se a ocorrência 27.285 óbitos por doenças respiratórias. Ou seja, ainda sem influência do SARS-CoV-2. Com a pandemia, durante os anos de 2020 e 2021, as doenças respiratórias não-covid decaíram para apenas 21.171, uma estrondosa queda de 22,4%. Ou, se se quiser, em valor absoluto 6.114 pessoas.
Deste modo, se se juntasse a covid-19 às doenças respiratórias, então durante a pandemia (2020-2021) terão ocorrido em meio hospitalar um total de 34.008 mortes, o que contrasta com 27.285 óbitos no período 2018-2019.
Nesta medida, só por aqui, o impacte líquido da pandemia será muito menor do que propalado: morreu-se muito por uma nova doença, mas, como em consequência “desapareceram” doenças respiratórias que tinham um impacte letal relevante, o saldo não se mostra assim tão elevado.
Óbitos por doenças do aparelho respiratório registados no mês de Janeiro entre 2017 e 2022. Fonte: SNS. Cálculos e análise: PÁGINA UM.
Contas feitas, em meio hospitalar, o acréscimo líquido é de 6.723 óbitos. Mas atenção: na segunda parte da análise, amanhã, veremos que é redutor estar apenas a usar, para calcular o impacte líquido do SARS-CoV-2, apenas as doenças respiratórias.
Aliás, nos últimos meses, a evolução da mortalidade das doenças respiratórias tem sido absurdamente atípica. No ano passado, houve apenas um mês (Janeiro) em que se ultrapassou a fasquia das mil mortes por este grupo de causas. Em 2017 houve sete meses; em 2018 registaram-se oito, em 2019 foram seis, e em 2020 foram apenas três, curiosamente os do primeiro trimestre, ou seja, imediatamente antes e no mês da chegada da covid-19 a Portugal.
Ou seja, literalmente, a covid-19 “sufocou” uma importante parte das doenças respiratórias.
No passado mês de Janeiro – que já consta na base de dados do SNS –, por doenças respiratórias não-covid foram contabilizadas 632 mortes, um valor completamente irrisório para um mês de Inverno. A título comparativo, em Janeiro de 2017 registaram-se, em meio hospitalar, 2.169 mortes por doenças respiratórias, ou seja, cerca de três vezes mais.