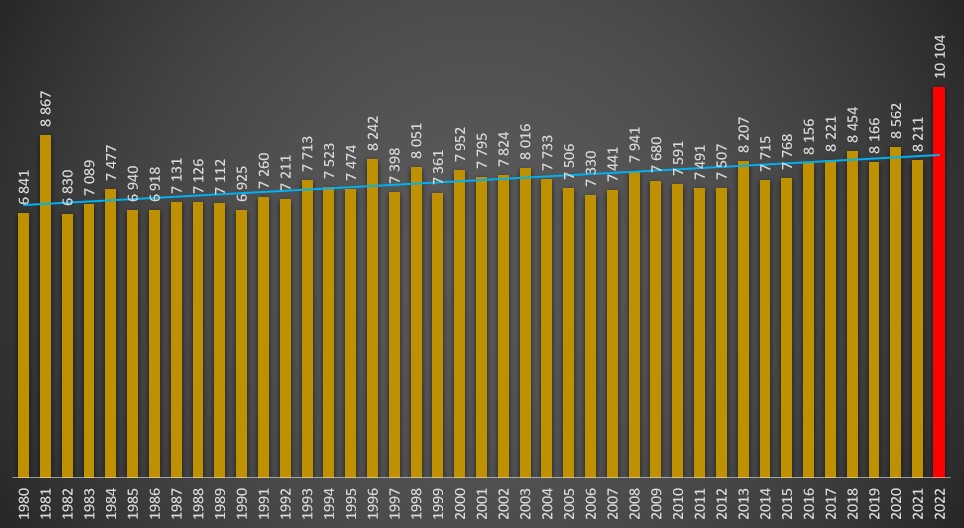
“Vai ficar tudo bem”, clamou-se nos primórdios da pandemia. Mas afinal está a “ficar tudo mal”, com a mortalidade por todas as causas a atingir valores absurdos para esta época do ano. Com base nos dados históricos, o PÁGINA UM estima que este mês, a poucas horas de terminar, houve um excesso de 60 óbitos todos os dias. Incluindo dias santos.
Um excesso de mortalidade de quase 1.800 óbitos – este será, para já, o saldo negro de um insólito mês de Junho em Portugal, de acordo com cálculos do PÁGINA UM baseados no histórico desde 1980 e dos valores expectáveis para esta época do ano.
Algumas horas antes de Junho encerrar, aquele que em situações normais deveria ser o segundo mês menos mortífero de um qualquer ano – apenas atrás de Setembro –, apresenta este ano valores de mortalidade total típicos de Inverno, quando o frio e os surtos de doenças do aparelho respiratório e circulatório fazem mais vítimas.
Segundo os dados disponibilizados em tempo real pelo Sistema de Informação dos Certificados de Óbito (SICO), até às 21 horas de hoje estavam já contabilizados 10.104 óbitos no mês de Junho. O valor deverá, contudo, chegar próximo das 10.200 mortes com as actualizações finais.
Este número contrasta com uma média de 8.323 óbitos no mês de Junho ao longo do último quinquénio (2017-2021), que englobam dois dos anos de pandemia. Nunca antes houve um registo estatístico de Junho acima sequer dos 9.000 óbitos. Por norma, apenas os meses de Janeiro, Fevereiro e Dezembro ultrapassam a fasquia dos 10.000 óbitos.
Antes do presente ano, o triste recorde em Junho pertencia a 1981. Há cerca de quatro décadas, uma extrema onda de calor entre os dias 9 e 21 de Junho – com termómetros a ultrapassarem os 43 graus em Beja, quase 42 em Lisboa e 39 no Porto – contribuiu fortemente para se atingir, no final desse mês, 8.867 óbitos. Só no dia 15 de Junho daquele ano foram registadas 676 mortes.
Mas nessa altura, na década de 1980, a população idosa não era tão numerosa como agora – e os óbitos rondavam geralmente, no sexto mês de cada ano, os cerca de sete mil.
Mais recentemente, mesmo com um tempo mais quente em Junho nos anos de 2013 e 2020, as mortes nunca ultrapassaram a fasquia dos 8.600 óbitos totais.
Porém, mesmo contabilizando o expectável incremento da mortalidade – devido ao incremento da população mais idosa –, a situação actual é deveras impressionante, ainda mais sabendo-se que, do ponto de vista de saúde pública, a Direcção-Geral da Saúde (DGS) andou parte do tempo mais preocupada com a varíola dos macacos, que não causou qualquer vítima mortal.
Óbitos totais no mês de Junho entre 1980 e 2022. Linha de tendência a azul. Fonte: INE e SICO. Análise: PÁGINA UM.
De facto, como em 2020 e 2021, os efeitos da pandemia não se fizeram sentir na transição da Primavera para o Verão, seria de esperar que a mortalidade total de 2021 rondasse os 8.300 óbitos, de acordo com a linha de tendência traçada pelo PÁGINA UM. Significa assim que os números de Junho deste ano são absurdamente elevados, sem que haja uma explicação oficial coerente.
No passado dia 17 de Junho, a Direcção-Geral da Saúde (DGS) alegou que a mortalidade mais elevada deste mês se devia à conjugação da covid-19 – num país com uma das mais elevadas taxas de vacinação do Mundo e com a maior prevalência de covid-19 desde o início da pandemia – com o ‘aumento da temperatura média do ar’. A entidade liderada por Graça Freitas dizia mesmo que “este indicador tem estado ‘acima do normal para esta época do ano’”, o que é desmentido pelos dados do Índice Ícaro constantes do Portal da Transparência.
Com efeito, ao longo de Junho, dos 30 dias destes mês, houve 22 com valor de zero neste índice – ou seja, sem qualquer impacte na mortalidade. E o valor máximo foi de apenas 0,11 no dia 13, o que não é excessivamente relevante. No dia 14, por exemplo, foi apenas de 0,06, mas isso não impediu que se tenham atingido os 421 óbitos, quando a média diária no último quinquénio é inferior a 280 mortes.
Recorde-se que a DGS, apesar de poder saber quais são as doenças que estiveram a matar mais do que seria suposto ao longo de Junho – recorrendo aos dados em bruto do SICO –, nada tem investigado sobre este excesso. Por sua vez, o Ministério da Saúde tem-se também oposto a que o PÁGINA UM aceda a essa base de dados para realizar uma análise independente, razão pela qual está em curso um processo de intimação no Tribunal Administrativo de Lisboa.
Com a espuma dos dias a desaparecer em redor da pandemia, começam a surgir investigadores com coragem para análises menos emotivas e mais científicas. Anteontem, na prestigiada BMJ Global Health foi publicado um extenso artigo de nove investigadores de diversas universidades dos Estados Unidos, Canadá e Reino Unido onde não se poupam críticas aos abusos cometidos na gestão da pandemia que colidiram “com os direitos humanos e promoveram a polarização social, afectando a saúde e o bem-estar”.
Nove investigadores norte-americanos, canadianos e britânicos acusam as políticas de vacinação contra a covid-19, seguidas pelos diversos países democráticos, de terem tido “efeitos prejudiciais na confiança do público, na confiança nas vacinas, na polarização política, nos direitos humanos, nas desigualdades e no bem-estar social”.
Num extenso artigo de 14 páginas publicado na passada quinta-feira na prestigiada revista científica BMJ Global Health, os nove investigadores – que trabalham, entre outros centros, na Universidade de Oxford, Johns Hopkins University (Maryland), London School of Hygiene & Tropical Medicine, Universidade de Washington e Universidade de Toronto – questionam “a eficácia e as consequências da política de vacinação coerciva na resposta à pandemia”, recomendando aos decisores políticos que “retomem abordagens de saúde pública não discriminatórias e baseadas na confiança.”
Intitulado “The unintended consequences of COVID- 19 vaccine policy: why mandates, passports and restrictions may cause more harm than good”, o artigo aborda, em detalhe, como foi implementada a estratégia de vacinação maciça e as suas implicações em termos de psicologia comportamental (reactância, dissonância cognitiva, estigma e desconfiança), política e direito (efeitos nas liberdades civis, polarização e governança global), socio-economia (efeitos na desigualdade, capacidade do sistema de saúde e bem-estar social) e de integridade da Ciência e da Saúde pública (a erosão da ética da saúde pública e da supervisão regulatória). E também a forma ziguezagueante como políticos e media se comportaram.
Reconhecendo que as vacinas tiveram impacto significativo na redução da taxa de mortalidade relacionada com a covid-19, os investigadores criticam sobretudo os mecanismos de coerção e estigmatização implementados nos últimos dois anos, que “provocaram considerável resistência social e política”, o que, segundo eles, tiveram “consequências prejudiciais não intencionais”, as quais “podem não ser éticas, cientificamente justificadas e eficazes.”
Primeira página do artigo.
Por exemplo, no caso da adopção dos certificados digitais, como passes sanitários para o acesso a determinados locais, os investigadores salientam que acabou por “colidir com os direitos humanos e promover a polarização social afectando a saúde e o bem-estar”, tendo sido usado com um fito “inerentemente punitivo, discriminatório e coercitivo.” Defendem, por isso, ser da máxima importância uma reavaliação “à luz das consequências negativas.”
No artigo relembra-se também a manipulação da opinião pública em redor da eficácia das vacinas ao longo do ano passado para incentivar a adesão da população.
“A lógica comunicada publicamente para a implementação de tais políticas mudou ao longo do tempo”, salientam os autores. Numa primeira fase dizia-se que a vacinação visava a “proteção dos mais vulneráveis”. Em seguida serviria para se alcançar a “imunidade de rebanho’, acabar com a pandemia’ e ‘voltar ao normal’, assim que o suprimento de vacinas fosse suficiente”. Porém,“no final do Verão de 2021” já passou a defender-se “a recomendação universal de vacinação para reduzir a pressão hospitalar e nas unidades de cuidados intensivos na Europa e América do Norte”.
Sobre as políticas gerais da vacinação obrigatória, os autores admitem que têm sido cada vez mais desafiadas e questionadas, devido à diminuição significativa da eficácia contra a infecção, apontando também que estudos realizados em Israel e no Reino Unido mostram que a “vacinação forçada aumentou os níveis de contestação, especialmente naqueles que já desconfiavam das autoridades”, agudizando a polarização social.
Neste aspecto, os media mainstream são particularmente criticados pelos investigadores, por terem usado “narrativas simplistas sobre percepções públicas complexas”, sobretudo quando sistematicamente optaram por catalogar as posições críticas como uma “consequência de forças ‘anti-ciência’ e de ‘extrema-direita”.
Nessa linha, a pressão social sobre os não-vacinados chegou a níveis de perseguição. Por exemplo, ainda que a imunidade natural – adquirida por uma infeção anterior por SARS-CoV-2, tenha fornecido uma protecção significativa, mesmo superior à da imunidade vacinal, “muitos dos que foram infetados acabaram por ser suspensos dos seus empregos ou até mesmo despedidos”, no caso de não se terem vacinado, denunciam os investigadores. “Estas pessoas, ficaram impedidas de viajar ou de participar em eventos públicos”, acrescentam.
Não ser vacinado passou a ser alvo de uma discriminação automática, incentivada por políticos e mesmo pelos media. Discriminar ou rotular não-vacinados “tornou-se socialmente aceitável entre os grupos de pró-vacinas, media e o público em geral, que viram a vacinação completa como uma obrigação moral e parte do contrato social”, referem os investigadores, mas apontam as consequências nefastas: “O efeito, no entanto, tem sido o de polarizar a sociedade – física e psicologicamente (…) A política de vacinas parece ter impulsionado as atitudes sociais em direção a uma dinâmica nós/eles em vez de adaptativa com estratégias para diferentes comunidades e grupos de risco.”
Para exemplificar, as atitudes hostis de responsáveis políticos, os investigadores elencam frases ameaçadoras e estigmatizantes de diversos políticos, como Emmanuel Macron, Justin Trudeau, Joe Biden, Jacinda Ardern e Tony Blair.
A declaração do presidente francês, feita no início de Janeiro deste ano, é bastante reveladora da procura de estigmatização: “É uma pequena minoria que está a resistir. Como reduzir essa minoria? Irritando-os ainda mais… Quando a minha liberdade ameaça a liberdade dos outros, eu passo a ser um irresponsável e alguém irresponsável não é um cidadão”.
Também a de Tony Blair é destacada: “Precisamos chegar aos não-vacinados. Francamente, se você ainda não está vacinado, se é elegível e não tem razões de saúde para não ser vacinado, você não é apenas um irresponsável, mas um idiota.” E também são salientadas duas intervenções do presidente norte-americano, uma das quais em Setembro do ano passado em que responsabilizava os não-vacinados pela manutenção da pandemia. Joe Biden garantia que se estava perante uma “pandemia de não-vacinados”. Como agora se sabe, as vacinas concedem uma protecção extremamente curta ou mesmo irrelevante na redução da transmissão.
Segundo os investigadores, “os governos abusaram [também] do poder, invocando um constante estado de emergência, evitando [assim] a consulta pública”, além de terem demonstrado “que confiavam excessivamente nos dados fornecidos pelas farmacêuticas”.
Considerando também que “a confiança nas autoridades de saúde se perde quando estas não são transparentes” – até porque não existiu transparência sobre o impacto negativo das vacinas, o que “exacerbou as ansiedades sociais, frustrações, raiva e incerteza”, os investigadores concluem que “as consequências criadas por estas circunstâncias, provocam uma tensão entre os princípios constitucionais e bioéticos, especialmente em democracias liberais”. Razão que os leva depois a relembrar que “as estruturas éticas foram projetadas para assegurar que os direitos e liberdades sejam respeitados mesmo durante a emergência de saúde pública”.
António Morais acumula a presidência da Sociedade Portuguesa de Pneumologia (SPP) com as funções de consultor da Direcção-Geral da Saúde e do Infarmed. Lei diz que só poderia acumular se a SPP recebesse das farmacêuticas no máximo 50.000 euros por ano em média no quinquénio anterior. A SPP recebeu no período 2017-2021 cerca de 870 mil euros, ou seja, 17 vezes mais.
A Inspecção-Geral das Actividades em Saúde (IGAS) está a investigar o presidente da Sociedade Portuguesa de Pneumologia, António Morais. A abertura formal de um “Processo de Esclarecimento, o qual se encontra em curso” foi admitida pelo inspector-geral desta entidade, António Carapeto, em carta a que o PÁGINA UM teve acesso.
De acordo com o IGAS, um processo de esclarecimento deste tipo constitui um “procedimento rápido e expedito destinado à recolha de elementos com vista ao esclarecimento de expediente geral, à verificação prévia de requisitos que habilitem a eventual decisão de instauração de acção inspectiva ou ao acompanhamento de acções inspectivas em curso dentro ou fora” desta entidade.
António Morais (ao centro), preside à Sociedade Portuguesa de Pneumologia, e é consultor da DGS e do Infarmed.
Na base da abertura desta investigação está a notícia do PÁGINA UM de 18 de Abril passado que denunciou que António Morais está a violar há três anos, desde que tomou posse como presidente da SPP, as regras de incompatibilidade que o deveriam impedir de se manter como consultor do Infarmed e da Direcção-Geral da Saúde. As decisões administrativas que tenham sido tomadas com base em pareceres em que este pneumologista tenha participado são juridicamente nulas.
António Morais – que desde 2016, e apresenta-se como tal no seu currículo, é consultor de doenças intersticiais pulmonares do Programa Nacional para as Doenças Respiratórias da Direcção-Geral da Saúde (DGS) e membro da Comissão de Avaliação de Tecnologias de Saúde do Infarmed – não poderia estar a acumular aquelas funções públicas com as de membro dos órgãos sociais de uma sociedade profissional com tão estreitas relações comerciais com farmacêuticas.
Um decreto-lei de 2014 estipula que consultores, membros de comissões, grupos de trabalho e júris de concursos com determinadas funções em organismos do Ministério da Saúde não podem ser, em simultâneo, membros de órgãos sociais de sociedades científicas – como é o caso da SPP – que “tenham recebido financiamentos de empresas produtoras, distribuidoras ou vendedoras de medicamentos ou dispositivos médicos, em média por cada ano num período de tempo considerado até cinco anos anteriores, num valor total superior a 50.000”.
Ora, António Morais preside à SPP desde 14 de Janeiro de 2019, e esta sociedade médica ultrapassa larguissimamente o patamar dos 50 mil euros anuais. Quando este pneumologista – que exerce no Hospital de São João e na Trofa Saúde, além de ser também professor na Faculdade de Medicina do Porto – tomou posse, a SPP tinha recebido no quinquénio anterior uma média de 799.634 euros do sector farmacêutico, ou seja, 16 vezes mais do que o limite imposto pela norma das incompatibilidades.
No quinquénio 2017-2021, que engloba já os três anos de presidência de António Morais, os montantes arrecadados pela SPP ainda aumentaram mais: situaram-se nos 870.512 euros por ano. Para este aumento muito contribuiu o ano passado em que a SPP recebeu um financiamento recorde vindo do sector farmacêutico: 1.301.972 euros.
Em 2022, até ao dia de hoje, de acordo com a Plataforma da Publicidade e Transparência do Infarmed, a SPP amealhou 499.228 euros, mas usualmente a maior fatia de patrocínios e contratos comerciais com a indústria farmacêutica regista-se no último trimestre de cada ano no âmbito do Congresso de Pneumologia.
A título pessoal, António Morais tem também relações comerciais com farmacêuticas. Este ano já recebeu 10.281 euros provenientes de sete farmacêuticas.
Apoios do sector farmacêutico (em euros) à Sociedade Portuguesa de Pneumologia entre 2017 e 2021. Fonte: Infarmed.
Para além de questões éticas, as incompatibilidades de António Morais têm consequências legais e jurídicas muito graves. De acordo com o artigo 5º do Decreto-Lei nº 14/2014, “os pareceres emitidos ou as decisões tomadas por comissões, grupos de trabalho, júris e consultores, em que intervenham elementos em situação de incompatibilidade não produzem quaisquer efeitos jurídicos”, o que significa, em consequência, que “as decisões dos órgãos deliberativos (…) são nulas”, caso se baseiem naqueles pareceres.
António Morais, por seu turno, pode vir também a ser sancionado, porque o artigo 6º do mesmo diploma legal determina a obrigatoriedade de ele cessar as suas funções de consultor a partir do dia de tomada de posse como presidente da SPP (14 de Janeiro de 2019). O PÁGINA UM teve acesso à sua última declaração, com data de 5 de Março de 2018 – numa altura, portanto, em que ainda não presidia à SPP, e não estaria a violar o regime de incompatibilidades –, e que ainda consta no site do Infarmed.
Por essa falha, a IGAS pode, de acordo com a lei, aplicar-lhe uma coima entre 2.000 e 3.500 euros. Ou, simplesmente, não fazer nada, e a SPP continuar a receber aqueles montantes das farmacêuticas, tendo um presidente a aconselhar a DGS e o Infarmed como se fosse um perito independente.
A Ordem dos Médicos abriu mesmo um processo disciplinar a Jorge Amil Dias, presidente do Colégio da Especialidade de Pediatria, por delito de opinião, através de uma queixa de médicos com ligações à indústria farmacêutica. Amil Dias está obrigado a responder até ao final deste mês. Este é já o segundo processo disciplinar intentado contra este especialista pela Ordem dos Médicos durante o mandato de Miguel Guimarães. Sempre por delito de opinião.
A Ordem dos Médicos, dirigida pelo urologista Miguel Guimarães, decidiu mesmo dar provimento à queixa de 16 médicos – alguns dos quais com fortes ligações à indústria farmacêutica, como Filipe Froes, Carlos Robalo Cordeiro e Luís Varandas – contra Jorge Amil Dias, presidente do Colégio da Especialidade de Pediatria.
A queixa já foi “processada” pelo Conselho de Disciplina da Regional Sul da Ordem dos Médicos, presidida por Maria do Céu Machado, ex-presidente do Infarmed, e o PÁGINA UM teve conhecimento que a acusação foi já formulada com vista à aplicação de uma sanção. O pediatra Amil Dias tem até ao final deste mês para apresentar defesa.
O “crime” deste renomado especialista em gastroenterologia pediátrica é simples de explicar: durante a pandemia da covid-19, tomou posição pública, a título pessoal, ao considerar a vacinação de crianças “desproporcionada” e “desnecessária”, além de advogar a relevância da imunidade natural. Além disso, foi um dos subscritores de um abaixo-assinado que integrou quase uma centena de médicos e outros profissionais de saúde, alertando também para os riscos da vacinação num grupo etário de baixíssimo risco.
O processo disciplinar contra o presidente do Colégio de Especialidade de Pediatria – que não é escolhido, assim como nos outros colégios, nas mesmas eleições do bastonário, e beneficia de independência – resultou de uma carta-denúncia no início de Fevereiro, assinada por médicos afectos ao bastonário e à indústria farmacêuticas.
Miguel Guimarães, que se manifestou incomodado por pediatras contrariarem as suas posições de médico urologista a falar de assuntos de pediatria, anunciou mesmo que levaria o assunto a reunião do Conselho Nacional Executivo. O PÁGINA UM sabe, contudo, que nenhum efeito teria: aquele órgão da Ordem dos Médicos não tem poder para destituir membros de um Colégio da Especialidade.
Mais do que qualquer castigo relevante que possa atingir Jorge Amil Dias, este processo da Ordem dos Médicos revela o “clima de guerra” que alimenta as relações entre estes profissionais de saúde no mandato de Miguel Guimarães, que escancarou portas a procedimentos inquisitoriais por meros delitos de opinião, sobretudo com o advento da pandemia.
Miguel Guimarães tem sido, além disso, criticado internamente por não acatar os pareceres técnicos dos Colégios de Especialidade – e até de os esconder publicamente, razão pela qual o PÁGINA UM está a preparar um processo de intimação junto do Tribunal Administrativo –, optando antes por criar órgãos de consulta não-estatutários.
Um exemplo paradigmático foi o Gabinete de Crise contra a Covid-19, dirigido por Filipe Froes, um dos médicos portugueses com mais relações promíscuas com a indústria farmacêutica. Só este ano, Filipe Froes vai já em 18 mil euros recebidos deste sector, aproximando-se assim dos 400 mil euros declarados no Portal da Transparência e Publicidade do Infarmed desde 2013.
Miguel Guimarães (à direita), urologista e bastonário da Ordem dos Médicos, ao lado de Carlos Robalo Cordeiro, um dos subscritores da queixa contra Jorge Amil Dias.
Embora Miguel Guimarães continue sem impor um código de conduta, optando por rodear-se de médicos com ligações à indústria farmacêutica – e a própria Ordem dos Médicos recebeu, no ano passado, cerca de 430 mil euros deste sector –, a sua veia punitiva não tem deixado de latejar contra quem não segue a sua opinião.
Além deste processo contra Amil Dias, a Ordem dos Médicos intentou, durante a pandemia, diversos processos a membros do denominado grupo Médicos pela Verdade. Até mesmo Fernando Nobre, fundador da AMI e ex-candidato à Presidência da República, foi alvo de um processo disciplinar com proposta de sanção, estando actualmente em fase de recurso.
Mas mesmo antes da pandemia, durante o “reinado” de Miguel Guimarães, a Ordem dos Médicos começou a querer punir profissionais que simplesmente davam a sua opinião. Um exemplo, apurou o PÁGINA UM, é a carta-aberta, publicada no jornal Público em Outubro de 2019, de um conjunto de 10 pediatras, entre os quais também Jorge Amil Dias, que criticava a então situação problemática das urgências pediátricas.
Os centros de vacinação COVID (CVC) no Alentejo não dão alternativa imediata a quem não queira ser inoculado com doses de lotes que beneficiaram de extensão ad hoc do prazo de validade. Infarmed diz agora que houve autorização da Agência Europeia do Medicamento, mas não disponibiliza o documento nem apresenta justificação para o secretismo da medida. Ministra da Saúde mantém silêncio, não se sabendo sequer quantas pessoas foram vacinadas nestas condições nem sequer como e quem avaliará eventuais efeitos adversos da decisão de maximizar o uso de vacinas apenas para, supostamente, se poupar algum dinheiro.
Diversos Centros de Vacinação COVID (CVC) do Alentejo que estão a usar lotes de vacina fora de prazo de validade – cuja administração obteve uma autorização informal do Infarmed, através de um simples e-mail enviado em Março, conforme ontem divulgado pelo PÁGINA UM – estão a recusar uma alternativa imediata aos utentes que não queiram ser injectados nessas condições. Se recusarem, as pessoas não são vacinadas com outro lote, e ficam a aguardar convocatória em data incerta.
Contudo, não é certo que todos os utentes estejam a ser avisados, uma vez que o consentimento informado para a administração destas vacinas é meramente oral, sem comprovativo escrito sobre as condições das vacinas e efeitos adversos previsíveis apresentados de forma quantitativa.
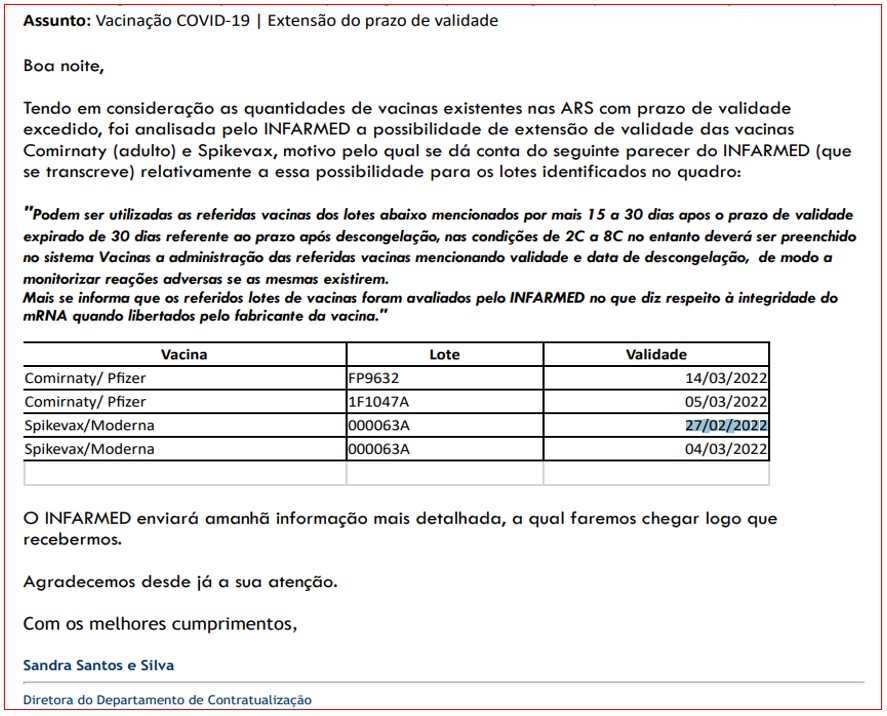
Em causa está assim um número indeterminado de doses pertencentes a três lotes específicos de vacinas contra a covid-19, e que receberam uma autorização ad hoc para continuarem a ser administradas após o prazo de validade. São os casos dos lotes FP9632 e 1F1047A da vacina Comirnaty/Pfizer (com prazo de validade até 14 de Março e 5 de Março, respectivamente), e ainda do lote 000063A da vacina Spikevax/Moderna. Para o lote desta segunda vacina, alguns frascos tinham expirado o prazo de validade em 27 de Fevereiro e outros em 4 de Março.
Em circunstâncias normais, os frascos destes lotes deveriam ser imediatamente destruídos após esgotar-se o prazo de validade, segundo as normas do “resumo das características do medicamento” – inseridas no Portal Infomed. Porém, em Março, através de uma simples comunicação por correio electrónico à Administração Regional de Saúde (ARS) do Alentejo, o Infarmed concedeu uma autorização de prorrogação do prazo de validade .
Essa autorização especial não consta, porém, em qualquer das habituais circulares informativas do Infarmed nem sequer foi introduzida, com identificação dos lotes em causa, no resumo das características do medicamento.
Ontem à noite, pelas 22:21 horas, o PÁGINA UM recebeu um esclarecimento do Infarmed – por “solicitação do gabinete de comunicação do Ministério da Saúde”, informando que “a Agência Europeia de Medicamentos (EMA) aprovou, este ano, a extensão do prazo de alguns lotes de vacinas contra a COVID-19, em condições de ultracongelação”, acrescentando ainda que “a extensão de prazo, de três meses e de seis meses aplica-se a todos os Estados-membros e tem efeitos retroactivos relativamente a lotes de injetáveis produzidos antes da aprovação.”
Em vez de uma circular informativa, que esclarecesse e justificasse a medida de prorrogação do prazo de validade, o Infarmed decidiu apenas comunicar por correio electrónico a sua decisão ad hoc à ARS do Alentejo. Ignora-se quantas doses já foram usadas dos lotes em causa.
No entanto, apesar de reiterado o pedido de indicação dos lotes autorizados, o Infarmed não respondeu. O PÁGINA UM tentou obter informação no site da EMA, mas sem sucesso. Existem, no entanto, autorizações especiais de prorrogação de prazo para certos lotes da vacina da Pfizer pelo National Health Service (NHS), do Reino Unido, mas nenhum dos lotes são aqueles que o Infarmed autorizou ad hoc para o Alentejo.
O Infarmed também não quis explicar os motivos para não ter sido produzida qualquer circular – como é habitual sempre que formalmente existe uma decisão do Conselho Directivo – sobre esta matéria.
No seu esclarecimento de ontem, o INFARMED diz apenas que a decisão de “utilização das vacinas” fora do prazo se baseou em “estudos de estabilidade apresentados pelos laboratórios”, mas não os enviou nem nunca os disponibilizou.
Após a recepção deste esclarecimento, o PÁGINA UM questionou ainda o Infarmed no sentido de saber se o regulador informou a ministra da Saúde sobre a decisão de administrar vacinas fora do prazo sob autorização “especial”. E questionou também o Infarmed sobre se não se estaria perante um “ensaio clínico” ilegal, porquanto, como o PÁGINA UM salientou ontem, nos e-mails entre o Infarmed e a Administração Regional de Saúde do Alentejo prevê-se a monitorização específica das pessoas injectadas com vacinas fora do prazo inicial de modo a avaliar posteriormente os efeitos adversos e a efectividade vacinal.
O PÁGINA UM também insistiu, junto dos três assessores de imprensa da ministra da Saúde, Marta Temido, para saber se o Governo tinha conhecimento deste expediente autorizado pelo Infarmed, para conhecer se outras ARS foram abrangidas, quantas pessoas tinham sido vacinadas com estes lotes e se esta estratégia será mantida. Não houve, até agora, qualquer resposta.
No Alentejo, foram administradas a um número indeterminado de pessoas vacinas contra a covid-19 fora do prazo. A decisão foi tomada no passado mês de Março em articulação entre o Infarmed e a Administração Regional de Saúde daquela região, mas sem base legal e contra as normas dos fabricantes. Apesar de garantir ser um processo seguro, o Infarmed acabou por estabelecer a obrigatoriedade de recolha de informação sobre as pessoas injectadas com estes lotes para posterior avaliação de eventuais acréscimos dos efeitos adversos ou de redução da efectividade vacinal. O PÁGINA UM revela os lotes das vacinas da Pfizer e da Moderna que foram injectadas já depois de expirar o prazo de validade. O Ministério da Saúde (ainda) não comentou se sabia desta decisão nem esclareceu (ainda) se houve mais lotes fora do prazo usados em outras regiões do país.
O Infarmed autorizou o uso de três lotes de vacinas contra a covid-19 fora do prazo de validade em centros de vacinação do Alentejo durante o mês de Março e Abril, em condições que aparentam um ensaio clínico não autorizado, que não cumpre os mínimos princípios éticos e de consentimento informado.
De acordo com mensagens electrónicas a que PÁGINA UM teve acesso, na noite de 14 de Março passado a directora do Departamento de Contratualização da Administração Regional de Saúde (ARS) do Alentejo, Sandra Silva, informou diversos responsáveis daquela região que “tendo em consideração as quantidades de vacinas existentes nas ARS com prazo de validade excedido”, o Infarmed tinha autorizado a sua utilização.
Em causa estava um número indeterminado de frascos dos lotes FP9632 e 1F1047A da vacina Comirnaty/Pfizer, com prazo de validade até 14 de Março e 5 de Março, respectivamente, e ainda um lote 000063A da vacina Spikevax/Moderna, sendo que alguns frascos tinham expirado o prazo de validade em 27 de Fevereiro e outros em 4 de Março.
No e-mail daquela noite, além de acrescentar que seria enviada no dia seguinte “informação mais detalhada”, Sandra Silva transcrevia o parecer do Infarmed, constituído somente por duas frases escritas em português algo macarrónico: “Podem ser utilizadas as referidas vacinas dos lotes abaixo mencionadas por mais 15 a 30 dias apos o prazo de validade expirado de 30 dias referente ao prazo após descongelação, nas condições de 2C a 8C no entanto deverá ser preenchido no sistema Vacinas a administração das referidas vacinas mencionando validade e data de descongelação, de modo a monitorizar reações adversas se as mesmas existirem. Mais se informa que os referidos lotes de vacinas foram avaliados pelo Infarmed no que diz respeito à integridade do mRNA quando libertadas pelo fabricante da vacina”.
E-mail enviado pela directora do Departamento de Contratualização da ARS do Alentejo na noite de 14 de Março passado, informando sobre a decisão do Infarmed.
No site do Infarmed não consta qualquer aviso sobre esta matéria. Sobre as condições de conservação das vacinas covid-19, a última actualização é de 3 de Fevereiro deste ano, onde nada consta sobre a possibilidade de alargamento do prazo de validade.
E no Portal Infomed, e no caso do resumo das características do medicamento da vacina Comirnaty/Pfizer, além de se elencar de forma exaustiva as exigentes condições de conservação, salienta-se que, após descongelação, “os frascos para injectáveis por abrir podem ser conservados durante um total de 10 semanas a uma temperatura entre 2 oC e 8 oC, nunca ultrapassando o prazo de validade (VAL) impresso”.
No caso especifico da Spikevax/Moderna, o resumo das características do medicamento no Portal Infomed vão no mesmo sentido: “Não utilize esta vacina após o prazo de validade impresso no rótulo após VAL [prazo de validade]. O prazo de validade corresponde ao último dia do mês indicado”.
Em todo o caso, nunca fazendo referência às indicações dos fabricantes, na manhã do passado dia 15 de Março, a directora de Inspecção e Licenciamentos do Infarmed, Maria Fernanda Ralha, enviou um e-mail para ARS do Alentejo, explicitando melhor a “autorização” concedida.
Na mensagem aquela responsável do Infarmed garantia que “os referidos lotes das vacinas Comirnaty adulta e Spikevax mantém-se estáveis assumindo-se que nenhuma das outras condições de conservação/transporte aprovadas foi excedida [e que] poderão eventualmente se manter , por mais 15-30 dias, para além da validade aprovada quando as vacinas forem mantidas entre 2ºC e 8ºC desde a sua descongelação” (sic).
Maria Fernanda Ralha sugeria também que, “pela natureza destas vacinas COVID-19 e pelos dados de estabilidade disponíveis para outros lotes”, não se antevia para estes lotes fora de prazo “problemas de segurança”, mas em seguida acrescentava que “há no entanto que estar atentos a eventuais notificações de reações adversas em utentes que receberão estas doses pelo que é recomendado o registo deste desvio às condições aprovadas na plataforma Vacinas”. (sic)
Nessa medida, esta responsável do Infarmed acabou por instruir os responsáveis da ARS do Alentejo para tomarem obrigatórios “procedimentos, tendo em conta a salvaguarda da saúde pública”, entre os quais o registo na plataforma Vacinas da data de descongelação e administração da dose da vacina fora de prazo, de modo a ser possível uma “futura avaliação da efetividade vacinal e eventuais questões de farmacovigilância decorrentes destes desvios”.
Ou seja, assumia subliminarmente que não existiam certezas sobre a inocuidade do prolongamento do prazo de validade nem tão-pouco se ficaria afectada a protecção vacinal.
Esta decisão do Infarmed e da ARS do Alentejo não se deveu a qualquer quebra de stock de vacina. Pelo contrário, tem sido a fraca adesão às doses de reforço, sobretudo da população abaixo dos 50 anos, que tem causado “sobras” e, portanto, risco de partes de lotes expirarem o respectivo prazo de validade. Tanto assim é que a responsável do Infarmed recomendou que “as vacinas descongeladas e cujo prazo de validade aprovado já foi ultrapassado devem ser usadas antes de vacinas descongeladas em qualquer uma das datas subsequentes e só quando terminar o stock existente se passe para as vacinas descongeladas noutros dias”. (sic)
O PÁGINA UM contactou Maria Fernanda Ralha, directora de Inpecção e Licenciamentos do Infarmed, que não quis fazer comentários sobre este assunto, alegando estar de férias e que todas as informações respeitantes às vacinas contra a covid-19 deveriam ser fornecidas pela Direcção-Geral da Saúde.
Também o gabinete da ministra da Saúde, Marta Temido, foi questionado sobre se tinha conhecimento desta decisão do Infarmed e da ARS do Alentejo, e sobre quantas pessoas tinham sido vacinadas com vacinas fora do prazo de validade. E também se questionou o Ministério da Saúde sobre se noutras regiões se tinha usado similar procedimento, e se sim, se este procedimento seria mantido no futuro. Não houve, até agora, qualquer reacção.
O PÁGINA UM perguntou ao Infarmed que “corpo estranho” foi detectado há três semanas num lote de vacinas contra a covid-19 da Moderna. E quis também aceder a todos os documentos enviados pela Agência Europeia do Medicamento desde 2020. A resposta chegou ontem: confidencial. Tudo no “segredo dos deuses”. Mais um caso para ser resolvido no Tribunal Administrativo, recorrendo ao FUNDO JURÍDICO do PÁGINA UM.
O Infarmed argumenta a existência de “um dever de confidencialidade” para não identificar o “corpo estranho” encontrado num lote da vacina Spikevax, da farmacêutica Moderna, no início deste mês.
Após ter sido divulgado pelo regulador do medicamento, em 10 de Abril passado, que a Agência Europeia de Medicamentos (EMA) e a Agência Espanhola do Medicamento e Produtos Sanitários (AEMPS) havia “um defeito de qualidade detetado no lote 000190A da vacina COVID-19 Spikevax, no fabricante situado em Espanha do laboratório Moderna, que era a presença de um corpo estranho no frasco da vacina”, o PÁGINA UM quis saber qual era, afinal, esse “corpo estranho”, de modo a aquilatar da gravidade do caso.
Em 12 de Abril,o PÁGINA UM solicitou ao Infarmed que disponibilizasse os documentos enviados pela EMA e pela sua congénere espanhola onde “conste a identificação, de forma clara e evidente, do “corpo estranho no frasco da vacina”, ou outra qualquer informação que evidencie que essa informação não foi apurada por qualquer entidade.”
Igualmente, foi pedida permissão, ao abrigo da Lei do Acesso aos Documentos Administrativos, para “o acesso, para eventual obtenção de cópia (analógica ou digital), de todo e qualquer documento administrativo na posse do Infarmed que tenha sido transmitido por carta normal (em papel), por mensagem de correio electrónico, por outro qualquer sistema digital escrito, sonoro ou audiovisual pela Agência Europeia de Medicamentos (EMA), e outras entidades internacionais homólogas do Infarmed, desde 2020 até à data”. Ou seja, o PÁGINA UM pretende, como constitucionalmente consagrado, investigar as actividades de regulação dos medicamentos em Portugal.
Em resposta ontem ao PÁGINA UM, o Infarmed alega que o regime jurídico dos medicamentos de uso humano (Decreto-Lei n.º 176/2006) “prevê um dever de confidencialidade que se traduz num regime especial em matéria de acesso a documentos administrativos”, incluindo dados “transmitidos pela Agência [EMA] ou pela autoridade competente de outro Estado Membro.”
O regulador português, presidido por Rui Santos Ivo, aponta mesmo para o disposto no n.º 2 do artigo 188º desse regime que diz serem “confidenciais os elementos apresentados ao Infarmed ou a estes transmitidos pela Agência ou pela autoridade competente de outro Estado membro, sem prejuízo do disposto no presente decreto-lei.”
Ora, entre aquilo que é o “disposto” neste diploma legal está um aspecto essencial: a protecção da saúde pública. No artigo 4º salienta-se que “as disposições do presente decreto-lei [e, nessa medida, a questão da confidencialidade] devem ser interpretadas e aplicadas de acordo com o princípio do primado da protecção da saúde pública.”
Esse mesmo primado leva à necessidade do Infarmed publicitar as informações “na página electrónica”, conforme previsto no artigo 198º, casos como este (detecção de anomalias em medicamentos), mas não se concede o direito de sonegar elementos relevantes como seja a identificação do “corpo estranho” apenas com o objectivo de proteger uma farmacêutica.
Face à recusa desta informação, e sobretudo por revelar um procedimento anti-democrático e não-transparente, o PÁGINA UM irá apresentar mais um – o segundo – processo de intimação contra o Infarmed junto do Tribunal Administrativo, através do FUNDO JURÍDICO.
Direcção-Geral da Saúde eliminou frases comprometedoras sobre o remdesivir na norma terapêutica aprovada em Janeiro passado, e mantém o fármaco da Gilead como terapia possível no tratamento contra a covid-19. Portugal é o país europeu com mais pessoas que sofreram efeitos adversos pelo uso deste fármaco. Infarmed continua sem ceder dados detalhados pedidos pelo PÁGINA UM, que aguarda entretanto decisão do Tribunal Administrativo de Lisboa ao processo de intimação.
A Direcção-Geral da Saúde (DGS) continua a incluir a administração de polémico anti-viral remdesivir, produzido pela Gilead Sciences sob a marca Veklury, na terapêutica para a covid-19, mesmo conhecendo-se, cada vez mais, as evidências de graves efeitos adversos, e de o seu uso já ter sido abandonado pela esmagadora maioria dos países europeus.
Na revisão da Norma 004/2020, que rege as terapêuticas, e que entrou em vigor no sábado passado, dia 23 de Abril, o remdesivir ainda permanece – dir-se-ia, estoicamente – na lista de medicamentos para o tratamento de “pessoas internadas por pneumonia por SARS-CoV-2 e hipoxemia confirmada”, mas já apenas como uma alternativa a ser considerada após a dexametasona e o metilprednisolona.
Gilead conseguiu vender largas dezenas de milhões de euros em remdesivir para combate à covid-19 sem existir garantia de eficácia nem de segurança.
Mesmo assim a última revisão da norma “ameniza” uma actualização feita em Janeiro último, que era bastante comprometedora para o fármaco. Com efeito, na actualização de 5 de Janeiro, a referência ao fármaco da Gilead como opção secundária era acompanhada pela seguinte nota: “Até ao momento, o remdesivir não revelou benefício inequívoco ao nível da mortalidade avaliada aos 28 dias nos ensaios clínicos. Assim, a sua prescrição deve decorrer de uma avaliação clínica individualizada, com ponderação dos riscos e benefícios para o doente, e de acordo com o Resumo das Características do Medicamento (RCM).”
Estas duas frases foram agora eliminadas, sem qualquer justificação, e o medicamento continua a ser uma hipótese terapêutica.
Certo é que vai já longe o tempo em que o remdesivir chegou a ser de uso quase obrigatório contra a covid-19, podendo os médicos que não o prescreviam ter problemas se os doentes morressem, conforme admitiu recentemente em entrevista ao PÁGINA o antigo bastonário da Ordem dos Médicos José Manuel Silva.
Com efeito, em Outubro de 2020, quando este fármaco – mesmo sem ensaios clínicos cientificamente validados – começou a ser usado em Portugal, a Norma 004/2020 quase o tornou de uso obrigatório na abordagem terapêutica em regime de internamento.
O então ponto 40 dessa norma determinava que a “terapêutica com remdesivir deve ser administrada o mais precocemente” em doentes internados com “confirmação laboratorial de SARS-CoV-2” que apresentassem um quadro de pneumonia, saturação de oxigénio inferior a 94% e idade igual ou superior a 12 anos com peso igual ou superior a 40 quilogramas.
A contínua “sobrevivência” do remdesivir na Norma 004/2020 deve-se, quase em exclusivo, ao forte “lobby da Gilead” no interior da DGS e da Faculdade de Medicina de Lisboa, que tudo tem feito para não se assumir publicamente os efeitos adversos e sobretudo o desastre económico na sua aquisição. E isto muito fruto das promiscuidades políticas e médicas com a farmacêutica norte-americana.
Recorde-se que este medicamento, inicialmente prescrito, embora com fracos resultados, para o vírus ébola, acabou por cair nas graças da Comissão von der Leyen na primeira fase da pandemia, em 2020. Em 8 de Outubro daquele ano, a Comissão Europeia decidiu assinar um acordo de compra conjunto que literalmente obrigou 36 países comunitários e extra-comunitários da Europa a adquirirem grandes quantidades de remdesivir à Gilead a preços exorbitantes. Para este “brinde” à Gilead, a Comissão Europeia garantiu um financiamento de 70 milhões de euros para a compra de 200 mil frascos de Veklury.
Para cumprir a parte portuguesa no negócio, logo em 23 de Outubro, a DGS assinaria um contrato com a Gilead com vista ao pagamento de um primeiro lote de 54.600 frascos. Custo total: 19.458.000 euros, ou seja, 356 euros por unidade. Note-se que em Novembro de 2020, o Le Monde destacava que, apesar de o custo de produção do remdesivir atingir apenas 0,93 dólares por dose – o que implicaria um custo de 5,58 dólares por tratamento –, a farmacêutica vendia-a por um preço 420 vezes superior.
Portugal deveria ter ainda adquirido um segundo lote ao longo de 2021 no valor de 15.018.645 euros – conforme determinava uma Resolução do Conselho de Ministros assinada exclusivamente por António Costa –, mas por razões nunca explicada pela DGS e pela Gilead, apesar das perguntas do PÁGINA UM, apenas foi assinado um contrato em 12 de Julho do ano passado por um valor simbólico: um pouco menos de 16 mil euros.
Não deve ter sido, contudo, indiferente para este desfecho o desaconselhamento sobre o remdesivir feito ainda em Novembro de 2020 pela Organização Mundial de Saúde (OMS); apesar de uma recente actualização ter passado a recomendá-lo para pessoas não internadas, e nos Estados Unidos tenha sido aprovado pela FDA o seu uso em crianças com mais de três anos também não internadas, desde esta semana. Fracas vantagens (um benefício de custo económico extremamente elevado para quem não apresenta um quadro clínico sequer moderado) que não faz esquecer os efeitos adversos relevantes.
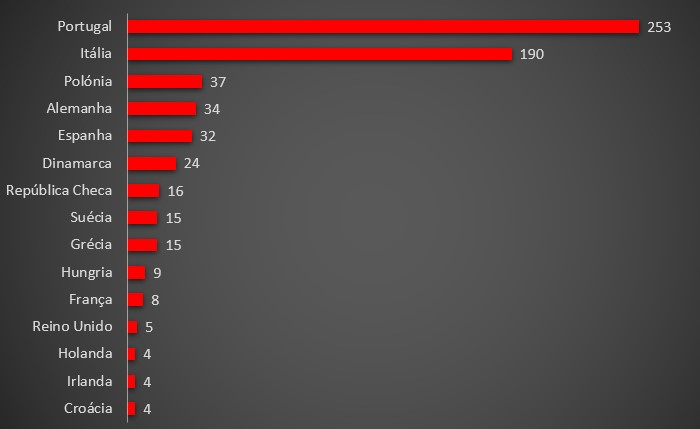
Com efeito, apesar do Infarmed continuar a recusar facultar dados detalhados sobre as reacções adversas em doentes-covid em Portugal, através do sistema EudraVigilance – base de dados agregada da Agência Europeia do Medicamento – observa-se que Portugal lidera o número absoluto de casos individuais com efeitos adversos causados pela administração de remdesivir, contabilizando-se já 253. Ignora-se quantos resultaram em mortes.
Número total de casos individuais com efeitos adversos ao remdesivir. Fonte: EudraVigilance.
O segundo país com mais casos é a Itália, com 190, mas com uma população seis vezes superior a Portugal. Casos adversos relacionados com o polémico fármaco da Gilead são relativamente escassos nos outros países da União Europeia.
O terceiro país com mais casos é a Polónia, apenas com 37, mas com uma população quase quatro vezes superior à portuguesa. A Alemanha contabilizou até agora 34 casos e tem mais de oito vezes a população portuguesa, enquanto a Espanha (com 46 milhões de habitantes) contou 32 doentes com problemas decorrentes do uso de remdesivir.
Em Portugal, o remdesivir sempre mereceu um carinho especial por parte dos denominados “peritos” que aconselharam a DGS nas terapêuticas para os doentes com covid-19.
De entre esses, destacam-se três médicos – Filipe Froes, António Diniz e Fernando Maltez – que simultaneamente integram a equipa de consultores da DGS para a elaboração e actualizações da Norma 004/2020 e sentam-se à mesa com a Gilead, e especificamente para falarem do remdesivir, uma vez que constam do seu advisory board desde 2020. Já este ano, Fernando Maltez e Filipe Froes receberam, cada um, 1.832,7 euros a esse título – pelo menos essa é a verba por eles declarada no Portal da Transparência e Publicidade do Infarmed.
Mas também a Faculdade de Medicina da Universidade de Lisboa tem sido uma forte aliada da Gilead na promoção do remdesivir. A Associação para Investigação e Desenvolvimento da Faculdade de Medicina (AIDFM) – presidida por Fausto Pinto, também director daquela instituição de ensino –, tem feito para a Gilead sucessivos estudos sobre este fármaco, mas que nunca viram a luz do dia.
Filipe Froes é um dos três consultores da DGS que se tem destacado na defesa do remdesivir. É também consultor da Gilead.
Durante o ano de 2020, a AIDFM recebeu desta farmacêutica 15.375 euros para um estudo intitulado “Análise do impacto de remdesivir na capacidade hospitalar do SNS” e mais 30.750 euros para o “Estudo de suporte do pedido de financiamento público de remdesivir no tratamento da covid-19”.
Já em 2021, encaixou mais verbas para o “Estudo comparativo sobre a utilização de remdesivir” (9.225 euros) e para a “Actualização do dossier de valor terapêutico de remdesivir (Veklury) na indicação aprovada” (12.300 euros). Este ano, no Portal da Transparência e Publicidade do Infarmed constam ainda mais dois estudos pagos pela Gilead: “Análise descritiva da utilização de remdesivir” (9.225 euros) e uma nova actualização do seu valor terapêutico (mais 12.300 euros).
A Gilead é, aliás, a farmacêutica com maior volume de negócios com esta associação da Universidade de Lisboa. Desde 2013, pelos mais diversos estudos e serviços, recebeu da farmacêutica norte-americana um total de 1.927.175 euros. Para se ter uma ideia da importância da Gilead nas contas da AIDFM, saliente-se que a segunda farmacêutica com maiores relações comerciais é a Bristil-Myers Squibb que “só” entregou 507.780 euros.
Entretanto, já este ano, a Gilead foi também “pescar” à política, contratando Ana Paula Martins, bastonária da Ordem dos Farmacêuticos até Fevereiro passado, e que acumulava com a docência na Faculdade de Farmácia de Lisboa. A agora directora de Assuntos Governamentais da Gilead é, desde Dezembro do ano passado, vice-presidente do Partido Social Democrata. Uma escolha de Rui Rio.
A Direcção-Geral da Saúde nunca divulgou um relatório sequer sobre os efeitos da pandemia nos lares, e procurou sempre falar pouco do assunto. O PÁGINA UM quis saber detalhes, e mais uma vez a entidade liderada por Graça Freitas recusou. A Comissão de Acesso aos Documentos Administrativos (CADA) diz agora, em parecer, que a DGS deve disponibilizar esses dados. Se a DGS persistir nesta postura, o PÁGINA UM accionará o seu FUNDO JURÍDICO para (mais) um processo de intimação junto do Tribunal Administrativo.
Em Novembro do ano passado, a Amnistia Internacional pediu ao parlamento italiano a realização de um inquérito independente sobre as mortes por covid-19 em lares de idosos e sobre “relatos de retaliação” que denunciaram condições de insegurança, relatou a Euronews.
Na província canadiana de Ontário, uma comissão de cuidados de longo prazo concluiu que a região “não estava preparada para uma pandemia e que as casas de repouso da província, que foram negligenciadas por décadas por sucessivos governos, eram alvos fáceis para surtos descontrolados”
No mês passado, um artigo no The Lancet destacava que a taxa de mortalidade nos lares ingleses em Abril de 2020 – quando surgiu a primeira vaga – tinha sido 17 vezes superior à registada nas pessoas com mais de 65 anos que viviam na comunidade, quando nos anteriores essa relação era de 10 vezes.
Também um artigo recente da Nature veio corroborar as críticas iniciais à gestão da pandemia da Suécia nos lares, embora de uma forma enviesada, porque comparou aquele país escandinavo apenas à vizinha Noruega, o país europeu menos afectado pela pandemia
Em Portugal, pouco relevo tiveram os efeitos da pandemia nos lares. Não houve estudos divulgados nem relatórios oficiais executados.
Sendo certo que foram frequentes as notícias de surtos e de mortes nas denominadas Estruturas Residenciais para Pessoas Idosas (ERPI), na verdade nunca houve um relatório oficial sobre o verdadeiro impacte da covid-19 naquela comunidade. As últimas informações oficiais quantitativas, mas nunca discriminadas por ERPI, foram dadas em 8 Fevereiro do ano passado quando a Direcção-Geral da Saúde (DGS) revelou que tinham morrido, até àquela data, 3.750 idosos em lares, das quais 42% entre 4 de Janeiro e 4 de Fevereiro. Esse número representava então 26% do total dos óbitos atribuídos à covid-19, numa altura em que os dados oficiais apontavam para 14.354 vítimas da pandemia.
Em Janeiro deste ano, o PÁGINA UM decidiu assim solicitar “ o acesso, para eventual obtenção de cópia (analógica ou digital), de todo e qualquer documento administrativo (em documento escrito ou sob a forma de base de dados) elaborado pela DGS, ou por outra entidade por sua iniciativa, ou ainda que esteja na sua posse, e que cont[ivesse] informação desde o início da pandemia, até ao momento da consulta, sobre o número de utentes, por Estrutura Residencial para Pessoas Idosas (ERPI), cujos óbitos [tivessem] ocorrido numa instituição com casos confirmados de covid-19 ou em utente ou trabalhador que [tivesse] apresentado sintomas compatíveis com a doença”.
Em suma, pretendia-se ter acesso às comunicações recebidas pela DGS, ou o suporte digital dessas comunicações após tratamento informático, em cumprimento do ponto 68 da Orientação nº 009/2020 de 11 de Março de 2020, com actualização em 10 de Janeiro deste ano.
Como habitualmente, a DGS não respondeu ao PÁGINA UM, mas um parecer da Comissão de Acesso aos Documentos Administrativos (CADA), aprovado na quarta-feira passada, considera agora que, apesar dos documentos solicitados respeitarem “a informação nominativa, reveste natureza quantitativa”, pelo que “livremente acessível”. E salienta que se a DGS quiser manter a recusa terá de “proferir decisão fundamentada” no prazo de 10 dias.
Caso a DGS não reconsidere a sua postura, este será mais um dos elementos a integrar num processo de intimação que o PÁGINA UM está já a preparar para entregar em breve no Tribunal Administrativo, recorrendo ao FUNDO JURÍDICO.
Saliente-se que este é um dos oito requerimentos à DGS apresentados pelo PÁGINA UM ao longo dos últimos meses sobre a pandemia – e que mereceram parecer da CADA. Além do pedido relacionado com os surtos e óbitos nas ERPI, o PÁGINA UM solicitou ainda o acesso às bases de dados dos internados e do Sistema Nacional de Vigilância Epidemiológica (SINAVE); a documentos sobre a evolução (temporal) da incidência cumulativa (real ou estimada) e as taxas de letalidade em Portugal das diferentes variantes do SARS-CoV-2 classificadas pela OMS como de preocupação (VOC) ou de interesse (VOI); os registos de surtos e mortes por covid-19 (como infecção nosocomial) em unidades hospitalares do SNS; os registos detalhados do conjunto de testes de deteção de SARS-CoV-2 e o número de casos positivos por idade ou faixa etária; os documentos produzidos no âmbito da actividade da Comissão Técnica de Vacinação contra COVID-19 (CTVC); e a consulta presencial do Sistema de Informação dos Certificados de Óbito (SICO).
Apenas no caso dos documentos da CTVC, a DGS satisfez, entretanto, parcialmente o requerimento do PÁGINA UM (cedendo os pareceres), mas ainda não disponibilizou as actas que, por exemplo, permitam identificar os consultores que votaram contra a vacinação dos adolescentes no Verão do ano passado. De entre os oitos pareceres, somente no caso do acesso ao SICO a CADA considerou que esta base de dados se encontra regida por uma legislação especial – negando assim razão ao PÁGINA UM –, questão que virá a ser dirimida nas instâncias judiciais.
A Direcção-Geral da Saúde garantiu, em Fevereiro passado, que cerca de 75% dos internados-covid tinham sido internados por consequência directa da infeção pelo SARS-CoV-2, mas quando o PÁGINA UM quis ver a base de dados com esses elementos, mais uma vez obteve o silêncio e o obscurantismo como resposta. A Comissão de Acesso aos Documentos Administrativos diz agora, em parecer, que a DGS deve disponibilizar a base de dados. Se a DGS persistir nesta postura, o PÁGINA UM accionará o seu FUNDO JURÍDICO para (mais) um processo de intimação junto do Tribunal Administrativo.
A Direcção-Geral da Saúde (DGS) deverá formalmente disponibilizar ao PÁGINA UM a base de dados dos doentes-covid desde o início da pandemia até à data, considera a Comissão de Acesso aos Documentos Administrativos (CADA) em parecer emitido por unanimidade em reunião na quarta-feira passada.
A deliberação da CADA, ontem enviada ao PÁGINA UM, resulta de mais uma recusa da DGS em divulgar documentos administrativos, desta feita a base de dados que a entidade liderada por Graça Freitas referiu existir quando, em comunicado de 4 de Fevereiro passado, divulgou que “no período entre 02/03/2020 e 10/12/2021, verifica-se que, das pessoas internadas com uma infeção por SARS-CoV-2, cerca de 75% estavam internadas por consequência direta dessa infeção.”
Nesse comunicado, a DGS confessava que “diariamente, as Administrações Regionais de Saúde recolhem manualmente de forma agregada junto dos hospitais o número total de camas ocupadas por pessoas com infeção por SARS-CoV-2/ COVID-19 em enfermaria e em Unidades de Cuidados Intensivos (UCI)”, acrescentando que “estes dados são comunicados à Administração Central do Sistema de Saúde (ACSS), que os processa e valida, partilhando-os posteriormente com a Direção-Geral da Saúde para divulgação.”
A existência da base de dados era, obviamente, conhecida pelo PÁGINA UM, tanto assim que tivera já acesso confidencial a uma cópia com registos individuais anonimizados entre Março de 2020 e meados de Maio, e que permitiu divulgar uma dezena de artigos de investigação inédita na comunicação social portuguesa durante a pandemia (Dossier P1 – Covid).
Nesta base de dados não existe qualquer informação pessoal que possibilite a identificação dos doentes, uma vez que todos os registos se encontram anonimizados, constando somente registos individualizados identificados com caracteres alfanuméricos, com indicação da idade, sexo, período e hospital de internamento, output (óbito ou alta), incluindo se esteve em unidade de cuidados intensivos, e comorbilidades e referências à evolução do estado clínico.
Aliás, no pedido do PÁGINA UM à DGS tinha sido salientado que os dados a disponibilizar deveriam ser anonimizados, para preservação da identidade e da esfera pessoal de cada doente. Cumprindo, aliás, não apenas o previsto no Regulamento Geral de Protecção de Dados como também o código deontológico dos jornalistas.
Com este pedido formulado pelo PÁGINA UM à DGS, em 21 de Fevereiro passado – que resultou, agora, no parecer da CADA –, pretendia-se sobretudo que esta entidade respeitasse o princípio de “arquivo aberto” da Administração Pública. Além disso, permitindo também uma actualização da informação – uma vez que o PÁGINA UM deixou de ter acesso à base de dados a partir de Maio do ano passado –, seria ainda possível confirmar, de forma independente, a veracidade e rigor das informações tornadas públicas pelo Governo e pela DGS.
A CADA, no seu parecer, destacando que “deverá ser cumprido o direito de acesso”, refere também que “se o volume ou complexidade da informação o justificarem, o prazo [de entrega dos documentos] (…) pode ser prorrogado até ao máximo de dois meses, devendo o requerente ser informado desse facto, com indicação dos respectivos fundamentos, no prazo de 10 dias”.
Saliente-se, contudo, que a base de dados consiste, num conjunto de elementos, que, apesar de numerosos, são susceptíveis de serem transpostos numa simples folha de cálculo (tipo Excel), de acesso imediato.
Caso não seja agora recebida finalmente uma cópia da base de dados dos internados-covid, este será mais um dos elementos a integrar num processo de intimação contra a DGS que o PÁGINA UM está já a preparar para entregar em breve no Tribunal Administrativo, recorrendo ao FUNDO JURÍDICO.
Saliente-se que este é um dos oito requerimentos à DGS já apresentados pelo PÁGINA UM ao longo dos últimos meses sobre a pandemia – e que mereceram outros tantos pareceres da CADA.
Além do pedido relacionado com a base de dados dos internados, o PÁGINA UM solicitou ainda o acesso à base de dados sobre surtos e óbitos em lares durante a pandemia; à base de dados do Sistema Nacional de Vigilância Epidemiológica (SINAVE); a documentos sobre a evolução (temporal) da incidência cumulativa (real ou estimada) e as taxas de letalidade em Portugal das diferentes variantes do SARS-CoV-2 classificadas pela OMS como de preocupação (VOC) ou de interesse (VOI); os registos de surtos e mortes por covid-19 (como infecção nosocomial) em unidades hospitalares do SNS; os registos detalhados do conjunto de testes de deteção de SARS-CoV-2 e o número de casos positivos por idade ou faixa etária; os documentos produzidos no âmbito da actividade da Comissão Técnica de Vacinação contra COVID-19 (CTVC); e a consulta presencial do Sistema de Informação dos Certificados de Óbito (SICO).
Apenas no caso dos documentos da CTVC, a DGS satisfez, entretanto, parcialmente o requerimento do PÁGINA UM (cedendo os pareceres), mas ainda não disponibilizou as actas que, por exemplo, permitam identificar os consultores que votaram contra a vacinação dos adolescentes no Verão do ano passado. De entre os oitos pareceres, somente no caso do acesso ao SICO a CADA considerou que esta base de dados se encontra regida por uma legislação especial – negando assim razão ao PÁGINA UM –, questão que virá a ser ainda dirimida nas instâncias judiciais.